Carbon dioxide là một sản phẩm dị hóa được tạo ra trong chu trình Krebs trong điều kiện oxi cung cấp cho tế bào bình thường. Là sản phẩm cuối cùng của quá trình hô hấp tế bào, các thông số có nguồn gốc carbon dioxide có thể được sử dụng để theo dõi tưới máu mô và phát hiện sự xuất hiện của quá trình chuyển hóa yếm khí trong trạng thái shock.
Trong bài viết này, chúng ta sẽ phân tích một số khía cạnh sinh lý, giá trị tiên lượng, ý nghĩa lâm sàng và các ứng dụng lâm sàng có thể có của sự khác biệt carbon dioxide tĩnh mạch với động mạch (Pv-aCO2) và tỷ lệ carbon dioxide tĩnh mạch – động mạch với chênh lệch oxy trong động mạch-tĩnh mạch (Cv-aCO2/Ca-vO2) trong trạng thái shock.
Tác giả: Thạc sĩ – Bác sĩ Hồ Hoàng Kim – ICU Bệnh viện NGUYỄN TRI PHƯƠNG
Shock là gì?
Shock là một tình trạng đe dọa tính mạng trong đó hệ thống tuần hoàn không thể cung cấp đủ oxy để duy trì nhu cầu trao đổi chất của các mô, dẫn đến rối loạn chức năng tế bào [1]. Do đó, việc nhận biết sớm quá trình giảm tưới máu mô và sự đảo ngược của nó là yếu tố then chốt trong việc hạn chế tiến triển thành rối loạn chức năng đa cơ quan và tử vong [2]. Các kỹ thuật hiện tại để theo dõi tưới máu mô chủ yếu tập trung vào lưu lượng máu toàn thân và sự cân bằng giữa nhu cầu oxy và cung cấp cho các mô [3, 4].
Trên thực tế, tối ưu hóa huyết động sớm bằng cách sử dụng các gói hồi sức nhắm vào bão hòa oxy tĩnh mạch trung tâm (ScvO2) và huyết động học vĩ mô ban đầu có liên quan đến việc giảm đáng kể tỷ lệ tử vong trong shock nhiễm trùng [5]. Tuy nhiên, tính hữu ích của các thông số có nguồn gốc từ oxy đã bị nghi ngờ mạnh mẽ [6], và các nghiên cứu gần đây đã không chứng minh được lợi ích lâm sàng của nó [7-9]. Trên thực tế, ScvO2 thường bình thường hoặc gần bình thường khi nhập viện ICU [10] và đạt được huyết động học vĩ mô bình thường và các thông số có nguồn gốc oxy toàn thân không loại trừ sự hiện diện hoặc tồn tại của tình trạng thiếu oxy mô. Trong bối cảnh này, các biến số khác như các tham số từ carbon dioxide (CO2) có thể cung cấp thông tin có giá trị về huyết động học vĩ mô và vi mô trong các giai đoạn đầu của cú shock, ngay cả khi các biến số oxy dường như đã được điều chỉnh [11-15]. Điều quan trọng là các biến số CO2 xảy ra nhanh hơn so với thay đổi nồng độ lactate, điều này tạo ra sự hấp dẫn của các thông số CO2 như là công cụ theo dõi trong giai đoạn đầu của quá trình hồi sức.
Nền tảng sinh lý
Sản xuất CO2 hiếu khí
Carbon dioxide (CO2) là một sản phẩm trao đổi chất cuối cùng được tạo ra trong điều kiện oxi cung cấp tế bào bình thường trong chu trình Krebs. Tổng sản lượng CO2 (VCO2) liên quan trực tiếp đến mức tiêu thụ oxy toàn thân (VO2) theo mối quan hệ, VCO2 = RQ × VO2 , trong đó RQ đại diện cho chỉ số hô hấp. RQ này phản ánh tỷ lệ mol CO2 được tạo ra trên mỗi mol oxy tiêu thụ ở cấp độ mô và nó sẽ thay đổi từ 0,6 đến 1 theo điều kiện trao đổi chất và chất nền năng lượng chiếm ưu thế. Do đó, VCO2 hiếu khí sẽ tăng hoặc trong quá trình chuyển hóa oxy hóa tăng (tức là, với sự tăng VO2 đồng thời) hoặc khi ở VO2 không đổi; chế độ ăn kiêng được thay thế bằng một lượng carbohydrate cao [16]. Trong điều kiện nghỉ ngơi hiếu khí, RQ sẽ không bao giờ > 1.0 vì sản xuất CO2 không được vượt quá lượng O2 tiêu thụ. Tuy nhiên, trong quá trình hoạt động cơ bắp mệt mỏi hoặc trong các tình huống bệnh lý nhất định, việc tạo CO2 yếm khí có thể chiếm tỷ lệ VCO2/VO2 > 1.0. Tuy nhiên, bất kể cơ chế tăng VCO2 hiếu khí, Pv-aCO2 sẽ chỉ tăng khi tăng bù trừ tim không đủ để làm sạch CO2 do các mô tạo ra.
Sản xuất CO2 yếm khí
Khi thiếu oxy mô, VCO2 hiếu khí giảm, trong khi VCO2 kỵ khí bật ra và tăng lên. Tăng VCO2 kỵ khí là hậu quả cuối cùng của quá trình đệm proton [H+ ] bởi bicarbonate bào tương và huyết tương (HCO3 − ). Sự “phóng thích H+ thô” được quan sát thấy trong quá trình thiếu oxy từ tổng số tất cả các phản ứng của tế bào giải phóng H+ (ví dụ: ATPase, các phản ứng hexokinase [HK], phosphofructokinase [PFK] và glyceraldehyd- 3-phosphate dehydrogenase [G3PDH]), đó là đối trọng bởi các phản ứng trao đổi chất tiêu thụ H + (ví dụ: các phản ứng creatine kinase [CK], AMP deaminase [AMPDase], pyruvate kinase [PK] và lactate dehydrogenase [LDH]). Do đó, sự khác biệt giữa phóng thích H+ thô và các phản ứng hóa học tiêu thụ H + (như là bộ đệm chuyển hóa) sẽ dẫn đến việc “phóng thích H + có tính hệ thống”, mà cuối cùng sẽ được điều chỉnh bởi hệ đệm cấu trúc nội và ngoại bào (ví dụ: axit amin) và hệ thống đệm bicarbonate [17]. Điều này sau đó là nguyên nhân chính cho sự gia tăng VCO2 kỵ khí, khi HCO3 − thu được H+ dư để trở thành H2CO3 và sau đó phân tách thành CO2 và H2O. Một nguồn bổ sung VCO2 kỵ khí là kết quả của quá trình khử carboxyl kỵ khí của một số chất như α-ketoglutarate và oxaloacetate xảy ra trong quá trình chuyển hóa trung gian, nhưng đóng góp của nó vào tổng VCO2 là khá nhỏ [18].
Mặc dù có tầm quan trọng về mặt sinh hóa, nhưng việc chứng minh lâm sàng về tăng CO2 kỵ khí có thể rất khó khăn vì tổng VCO2 giảm trong điều kiện thiếu oxy và lưu lượng máu tĩnh mạch có thể đủ để rửa sạch tổng CO2 được tạo ra tại các mô, do đó che lấp phần tăng CO2 kỵ khí .
Vận chuyển CO2 trong máu
Sự bài tiết carbon dioxide là một hiện tượng thụ động trong đó CO2 được chuyển xuống theo một độ chênh điện hóa từ các tế bào ra môi trường. Hiệu quả của việc vận chuyển này là một chức năng của quy ước (lưu lượng máu) và khả năng của chất vận chuyển (hàm lượng máu). May mắn thay, sự tiến hóa đã dẫn đến việc vận chuyển một lượng lớn CO2 trong máu mà không có sự thay đổi lớn trong lưu lượng máu. Carbon dioxide hòa tan gấp khoảng 20 – 30 lần so với oxy, nhờ đó CO2 hòa tan đóng vai trò chính trong tổng lượng vận chuyển của nó. Là một phân tử ái mỡ, CO2 nhanh chóng khuếch tán qua lớp lipid kép của tế bào và hồng cầu để được hydrat hóa và cuối cùng được chuyển đổi thành HCO3 − và H + . Do đó, nói chung, máu mang cả CO2 và các hợp chất liên quan của nó ở năm dạng:
- CO2 hòa tan: [CO2] DIS tuân theo định luật Henry, trong đó xác định rằng, ở nhiệt độ không đổi, bất kỳ chất khí nào hòa tan trong pha lỏng tỷ lệ với áp suất riêng phần của nó trong pha khí, được điều chỉnh bởi hệ số hòa tan khác với khí này . Trong điều kiện bình thường, ~ 5% tổng lượng CO2 được vận chuyển dưới dạng [CO2] DIS. Mặc dù có dung tích tương đối thấp trong máu, [CO2] DIS có vai trò quan trọng trong việc vận chuyển khí vì nó có thể nhanh chóng đi qua nội mô mạch máu, trong khi các dạng CO2 khác phải được chuyển đổi thành CO2 tự do để đi vào hoặc rời khỏi máu.
- Axit cacbonic: [H2CO3] là kết quả của phản ứng giữa CO2 và H2O. Ở mức độ pH của hầu hết các chất lỏng sinh lý, H2CO3 ngay lập tức phân tách thành H+ và HCO3 − . Do đó, [H2CO3] chỉ đại diện cho phần 1/400 của [CO2], theo đó, điều này không quan trọng về mặt định lượng đối với tổng lượng CO2 vận chuyển.
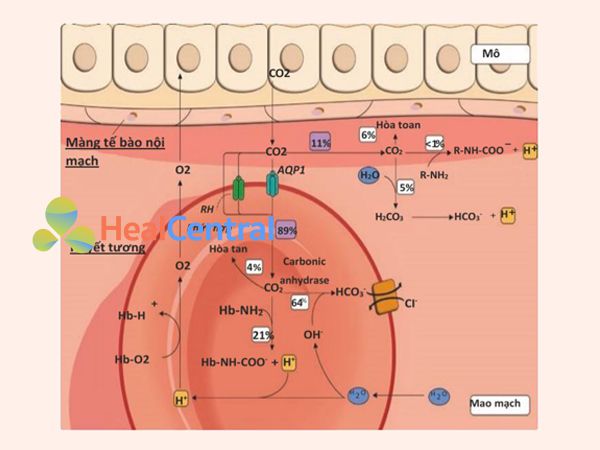
Hình 16.1: Các sự kiện nội bào và ngoại bào của vận chuyển CO2 trong máu - Bicarbonate: [HCO3 − ] có thể hình thành theo ba cách – bằng cách phân ly H2CO3 thành H+ và HCO3 − , bằng cách kết hợp trực tiếp CO2 và OH− (một phản ứng được xúc tác bởi anhydrase carbonic) và bằng cách kết hợp carbonate (CO3 2− ) và H+ . Trong máu động mạch, HCO3 − chiếm ~ 90% tổng lượng CO2. Do đó, CO2 kết hợp với nước (H2O) tạo thành axit carbonic (H2CO3) và chất này phân ly thành HCO3 − và ion hydro: CO2 + H2O = H2CO3 = HCO3 − + H+ . Carbonic anhydrase xúc tác gần như ngay lập tức phản ứng đầu tiên này chủ yếu xảy ra ở các tế bào hồng cầu (RBC) và các tế bào nội mô mao mạch phổi, trong khi phản ứng thứ hai không được xúc tác xảy ra với tốc độ chậm hơn nhiều. Khi H2CO3 phân tách trong RBC thành H+ và HCO3 − , H+ được đệm bởi hemoglobin, trong khi HCO3 − dư được vận chuyển ra khỏi RBC vào huyết tương bằng bộ trao đổi bicarbonate-chloride trung hòa điện tích (hình 16.1).
- Carbonate: [CO3 2− ] được hình thành chủ yếu từ sự phân ly bicarbonate: HCO3 − → CO3 2− + H+ . Do đó, [CO3 2− ] cao ~ 1/1000 so với HCO3 − ở pH 7,40. Theo đó, CO3 2− không quan trọng về mặt định lượng đối với việc vận chuyển CO2.
- Các hợp chất Carbamino: các nhóm protein không tích điện của protein có thể liên kết thuận nghịch với cả H + và CO2. Cho đến nay, hợp chất Carbamino quan trọng nhất là Carbamino hemoglobin (Hb-NH- COO− ), hình thành nhanh chóng và có thể đảo ngược khi CO2 phản ứng với nhóm amino tự do trên hemoglobin. Các hợp chất Carbamino chiếm ~ 5% tổng lượng CO2 trong máu động mạch.
Tổng hàm lượng CO2 (CCO2) trong máu động mạch là ~ 48 mL khí CO2/dL đo ở nhiệt độ tiêu chuẩn và áp suất/khô (STPD), tương ứng với PaCO2 là 40 mmHg. Từ 48 ml/dL đó, ~ 90% tương ứng với HCO3 − , trong khi hợp chất carbamino đóng góp với ~ 5%. Khi máu chảy dọc theo giường vi tuần hoàn, nó sẽ hấp thụ ~ 4 mL/dL CO2, do đó tổng CCO2 trong máu tĩnh mạch hỗn hợp sẽ tăng lên ~ 52 mL/dL. Từ CCO2 tăng dần đó, khoảng 10% tương ứng với CO2 hòa tan, ~ 69% với HCO3 − và ~ 21% với các hợp chất carbamino. Theo đó, các hợp chất CO2 và carbamino hòa tan rất quan trọng đối với việc mang CO2 gia tăng vào phổi do đóng góp của chúng vào sự gia tăng tổng lượng CO2 trong máu tĩnh mạch. Khi quá trình trao đổi chất oxy hóa xảy ra và chu trình Krebs duy trì chức năng của nó, ty thể tạo ra CO2, giúp khuếch tán ra khỏi tế bào qua khoang ngoại bào, qua nội mô mao mạch và vào huyết tương. Gần 11% CO2 gia tăng vẫn còn trong huyết tương trong suốt quá trình đến phổi, trong khi ~ 89% đi vào các tế bào hồng cầu, ít nhất là ban đầu. Phần lớn ~ 11% CO2 tăng trong huyết tương sẽ lần lượt được vận chuyển dưới dạng CO2 hòa tan (~ 6%, xem xét hematocrit 40%), như HCO3 − (~ 6%) và một lượng nhỏ dưới dạng hợp chất carbamino. Phần còn lại ~ 89% lượng CO2 gia tăng đi vào các tế bào hồng cầu thông qua hai “kênh khí” khác nhau: aquaporin 1 và phức hợp Rh. CO2 nội hồng cầu này sẽ được vận chuyển dưới dạng CO2 cytosolic hòa tan (~ 4%), trong khi ~ 21% lượng tăng như vậy sẽ được vận chuyển dưới dạng các hợp chất carbamino của Hb (tức là CO2 liên kết với hemoglobin). Các hợp chất carbamino trong RBC quan trọng hơn nhiều so với các hợp chất được hình thành trong huyết tương vì nồng độ hemoglobin trong RBC cao hơn đáng kể (~ 33 gr/dL) so với albumin, globulin và các protein huyết tương khác (~ 7 gr/dL protein huyết tương ). Hơn nữa, ái lực của CO2 đối với huyết sắc tố vượt xa so với protein huyết tương chính. Ngoài ra, ái lực của hemoglobin đối với H+ và CO2 sẽ được điều chỉnh miễn là nồng độ O2 thay đổi khi máu đi vào vi tuần hoàn mô hoặc quay trở lại phổi. Lượng CO2 gia tăng còn lại trong RBC sẽ được biểu thị bằng HCO3 − (~ 64%) do hoạt động anhydrase carbonic đẩy nhanh quá trình chuyển đổi CO2 thành HCO3 − . Nếu không có hoạt tính enzyme như vậy, HCO3 − khó có thể được tổng hợp bên trong các hồng cầu trong thời gian vận chuyển ngắn của các hồng cầu dọc theo mao quản. Hơn nữa, bộ trao đổi Cl-HCO3 AE1 (bộ trao đổi anion 1) mang HCO3 − mới được tổng hợp ra khỏi tế bào, thúc đẩy hình thành HCO3 − hơn nữa. Hình 16.1 nối lại các sự kiện kết hợp giữa hồng cầu và huyết tương trong vận chuyển CO2.
Đường cong phân ly CO2
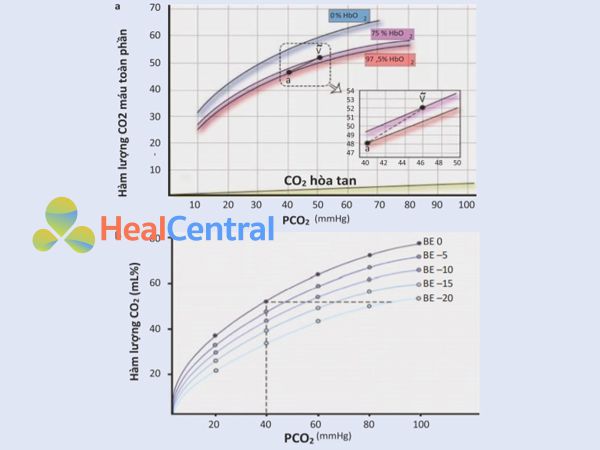
Việc vận chuyển tổng CO2 sẽ phụ thuộc vào pCO2, pH huyết tương và pO2 [19,20]. Đường cong phân ly CO2 được đặc trưng bởi mối quan hệ gần tuyến tính trong phạm vi sinh lý của các giá trị pCO2 và pO2 (Hình 16.2, bảng a). Hơn nữa, tại bất kỳ pCO2 nào, tổng hàm lượng CO2 tăng khi pO2 giảm. Kết quả là, khi máu đi vào vi tuần hoàn hệ thống và giải phóng O2, khả năng mang CO2 tăng lên, do đó máu có thể loại bỏ thêm CO2. Ngược lại, khi máu đi vào mao mạch phổi và liên kết với O2, khả năng vận chuyển CO2 sẽ giảm và máu mất khả năng vận chuyển thêm CO2. Do độ dốc đường cong phân ly CO2, pCO2 phải tăng từ 40 mmHg trong máu động mạch lên chỉ 46 mmHg trong máu tĩnh mạch hỗn hợp để tăng tổng hàm lượng CO2 thêm ~ 4 mL/dL (nghĩa là từ 48 đến 52 mL khí CO2/dL), cần thiết để loại bỏ CO2 được tạo ra bởi chức năng ty thể hiếu khí.
Hình 16.2. Đường cong phân ly CO2. Hình a, mối quan hệ tổng lượng CO2 trong máu toàn phần và áp suất riêng phần CO2 dựa theo sự biến thiên SpO2 (hiệu ứng Haldane). Hình b, ảnh hưởng của ion H+ trong tổng CO2 máu và áp suất riêng phần CO2.
Khác biệt CO2 máu tĩnh mạch – động mạch (Pv-aCO2)
Pv-aCO2 và mối quan hệ của nó với cung lượng tim
Sự khác biệt carbon dioxide từ tĩnh mạch đến động mạch (Pv-aCO2) đề cập đến độ dốc của áp lực riêng phần tạo bởi CO2 hòa tan trong máu tĩnh mạch hỗn hợp hoặc trung tâm và máu động mạch. Nhìn chung, Pv- aCO2 phụ thuộc vào tổng lượng carbon dioxide (CO2), cung lượng tim, mối quan hệ phức tạp giữa áp lực riêng phần CO2 và hàm lượng CO2 trong máu, và, có lẽ, phân phối lưu lượng máu vi tuần hoàn.
Phương trình Fick chỉ ra rằng sự đào thải CO2, tức là, tương đương với sản xuất CO2 (VCO2) ở trạng thái ổn định, nên bằng sản phẩm của cung lượng tim (CO) và chênh lệch CO2 của động mạch tĩnh mạch: VCO2 =CO x (CvCO2 – CaCO2).
Như đã đề cập ở trên, CCO2 và pCO2 duy trì mối quan hệ tương đối tuyến tính ở các phạm vi sinh lý thông thường. Do đó, các giá trị pCO2 đã được đề xuất như là chất thay thế cho CCO2 khi đánh giá sự khác biệt CO2 từ tĩnh mạch đến động mạch ở tại giường [20-23]. Kết quả là, một phương trình Fick được sửa đổi có thể thu được bằng cách thay thế pCO2 cho CCO2: ΔpCO2 = k x (VCO2 / CO).
Trong đó k là một hệ số giả tuyến tính được giả định là không đổi trong điều kiện sinh lý [22]. Tuy nhiên, trong điều kiện thiếu oxy nghiêm trọng, yếu tố k có thể tăng lên gấp sáu lần khi nhiễm toan chuyển hóa tăng, gây ra sự thay đổi trong mối quan hệ độ cong giữa CCO2 và pCO2 (Hình 16.2, bảng b). Do đó, yếu tố k tăng khi VCO2 giảm, nhưng hiệu quả của Pv-aCO2 sẽ phụ thuộc vào cung lượng tim và có thể phụ thuộc vào phân phối lưu lượng máu vi tuần hoàn.
Theo phương trình Fick đã sửa đổi, Pv-aCO2 và cung lượng tim giữ mối quan hệ đường cong ngược trong đó tăng Pv-aCO2 theo mức giảm dần của cung lượng tim, đặc biệt là ở các giá trị thấp hơn. Do đó, trong điều kiện ổn định của cả VO2 và VCO2, Pv-aCO2 tăng dần để đáp ứng với việc giảm cung lượng tim do hiện tượng “ứ đọng CO2” trong đó thời gian hồng cầu bị trì hoãn dẫn đến việc bổ sung CO2 cao hơn trong 1 đơn vị máu chảy qua vi mạch máu. Những quan sát ban đầu khi ngừng tim ở cả mô hình động vật và người cho thấy rõ mối liên hệ giữa làm chậm (hoặc ngừng) lưu lượng máu và tích lũy CO2 tĩnh mạch [24,25]. Tương tự, các mô hình thực nghiệm về xuất huyết, giảm thể tích máu và shock tắc nghẽn đã chứng minh mối quan hệ nghịch đảo này giữa Pv-aCO2 và cung lượng tim, do đó nhấn mạnh tầm quan trọng của sự ứ đọng dòng máu đối với sự tích tụ CO2 tĩnh mạch [26-29].
Tuy nhiên, tăng Pv-aCO2 ban đầu được hiểu là sự phản ánh tình trạng rối loạn sử dụng oxi mô vì các giá trị cung cấp oxy quan trọng dường như phù hợp với điểm mà CO2 tĩnh mạch bắt đầu tăng [26,27]. Trong một mô hình thí nghiệm trên chó bị tamponade tim bằng cách sử dụng nomogram Dill, Schlichtig và Bowles [30] đã đề xuất sự xuất hiện của VCO2 kỵ khí dưới điểm rớt DO2, do đó gợi ý mối liên hệ giữa rối loạn sử dụng oxi và tích lũy CO2 mô. Tuy nhiên, các mô hình thực nghiệm trong đó giảm dòng (máu) lũy tiến được sử dụng để đạt được việc cung cấp oxy tại điểm rớt (DO2) với mức giảm tiêu thụ oxy tiếp theo (VO2) có thể mang lại kết quả khó hiểu do không thể phân biệt được quá trình giảm tưới máu mô với rối loạn sử dụng oxy mô [31]. Để giải quyết vấn đề này, Vallet et al. [32] đã thiết kế một thí nghiệm để đo lường sự thay đổi Pv-aCO2 trong các chế phẩm máu từ chân sau của con chó được tách rời khỏi từ tuần hoàn hệ thống và kết nối với máy tuần hoàn ngoài cơ thể. Sự giảm tương đương của DO2 được tạo ra bởi hai cơ chế khác nhau của tình trạng thiếu oxy mô: một nhóm trải qua sự giảm dần lưu lượng máu bằng cách làm chậm tốc độ bơm của máy (tạo mô hình thiếu oxy máu do thiếu máu cục bộ), trong khi nhóm còn lại trải qua quá trình giảm pO2 động mạch thông qua giảm FiO2 (mô hình tạo rối loạn sử dụng oxy mô) nhưng bảo tồn tốc độ dòng chảy. Cả hai nhóm đều trải qua sự suy giảm tương tự ở DO2 và VO2, cho thấy mức độ rối loạn sử dụng oxy mô tương tự nhau. Tuy nhiên, Pv-aCO2 chân sau vẫn không đổi trong tình trạng thiếu oxy do thiếu oxy, trong khi nó cho thấy sự gia tăng hơn gấp đôi trong tình trạng thiếu oxy do thiếu máu cục bộ. Theo đó, các tác giả kết luận rằng “lưu lượng máu” là yếu tố chính quyết định Pv-aCO2, và do đó, việc không có Pv-aCO2 tăng lên không loại trừ sự hiện diện của chứng rối loạn sử dụng oxy mô.
Đánh giá một giả thuyết tương tự, Nevière et al. [33] đã so sánh tác động của giảm phân xuất oxy hít vào (thiếu oxy máu) so với giảm lưu lượng máu (thiếu oxy máu cục bộ) đối với sự khác biệt CO2 từ niêm mạc ruột đến động mạch (Pmtis-aCO2). Pmtis-aCO2 tăng tới 60 mmHg trong tình trạng thiếu oxy do thiếu máu cục bộ, trong khi nó vẫn gần như không đổi trong một loạt các giá trị DO2 trong tình trạng thiếu oxy. Thật thú vị, Pmtis-aCO2 tăng nhẹ khi sử dụng các giá trị FiO2 cực thấp. Các tác giả kết luận rằng sự gia tăng Pmtis-aCO2 chủ yếu được giải thích bằng sự thay đổi lưu lượng máu, mặc dù họ thừa nhận rằng pCO2 trong niêm mạc tăng trong điều kiện thiếu oxy nặng do giảm oxy máu có thể chỉ ra một số hình thành CO2 cục bộ. Tuy nhiên, thực tế là sự phụ thuộc DO2/VO2 đã đạt được sớm hơn so với sự gia tăng Pmtis-aCO2 trong điều kiện thiếu oxy do giảm oxy ngụ ý rằng Pmtis-aCO2 không nên được sử dụng như một dấu hiệu của chứng rối loạn oxi mô.
Tương tự, trong một mô hình thiếu oxy do xuất huyết mà không có hiện tượng giảm tưới máu trong đó mất máu tiến triển đã được thay thế bằng liều dextran với thể tích tương đương, Pv-aCO2 cho thấy không tăng khi lưu lượng máu được khôi phục, do đó khẳng định vai trò hàng đầu của lưu lượng máu đối với sự gia tăng CO2 trong máu tĩnh mạch [34].
Do đó, việc tăng Pv-aCO2 có liên quan mật thiết đến sự thay đổi cung lượng tim trong điều kiện không viêm. Tuy nhiên, sự phù hợp quan sát được giữa cung lượng tim và Pv-aCO2 trong shock nhiễm trùng là yếu [14, 35-37], điều này cho thấy các cơ chế khác có thể liên quan.
Pv-aCO2 và sự thay đổi dòng chảy trong vi tuần hoàn
Rối loạn chức năng vi tuần hoàn trong shock nhiễm trùng là một hiện tượng tổng quát được đặc trưng bởi giảm mật độ mao mạch chức năng (FCD-functional capillary density) liên quan đến việc gia tăng sự không đồng nhất của lưu lượng máu bao gồm các khu vực có mạch máu được tưới máu tốt gần với mao mạch không được tưới máu [38,39]. Trong điều kiện bình thường, sự không đồng nhất của lưu lượng máu vi tuần hoàn là không đáng kể [40], và sự phù hợp của tưới máu với chuyển hóa thường được cải thiện trong tình trạng thiếu oxy hoặc dòng chảy thấp [41]. Tuy nhiên, sự gia tăng tính không đồng nhất của lưu lượng máu vi tuần hoàn với việc giảm FCD sau đó có thể là nguyên nhân dẫn đến khả năng chiết xuất oxy bất thường xảy ra trong nhiễm trùng huyết [42,43]. Trên thực tế, việc ngừng dòng không đồng nhất của các mao mạch riêng lẻ có thể là một yếu tố quan trọng quyết định hiện tượng phụ thuộc vào việc cung cấp oxy trong các trường hợp shock nhiễm trùng nghiêm trọng nhất [42,44]. Điều quan trọng là sự thay đổi vi tuần hoàn có thể xảy ra ngay cả khi các thông số oxy hệ thống có vẻ đầy đủ, và nó dường như kích hoạt sự phát triển của rối loạn chức năng đa cơ quan [45]. Hơn nữa, các rối loạn vi tuần hoàn như vậy là yếu tố quyết định mạnh mẽ hơn đến kết quả lâm sàng so với các thông số huyết động vĩ mô, với sự gia tăng dần về nguy cơ tử vong ở các tứ phân vị cho các rối loạn nghiêm trọng nhất [46].
Mối liên hệ giữa sự thay đổi vi tuần hoàn và sự tích tụ CO2 của mô trong shock nhiễm trùng đã được đề xuất bởi Creteur et al. [47] bằng cách đánh giá đồng thời vi tuần hoàn dưới lưỡi, CO2 mô dưới lưỡi và CO2 niêm mạc dạ dày trong quá trình truyền liều dobutamine cố định liều ở mức thấp. Họ đã quan sát thấy sự tăng cung lượng tim và SvO2, trong khi khác biệt CO2 dưới lưỡi – động mạch (Psl-aCO2) giảm đáng kể (từ 40 ± 15 đến 17 ± 8 mmHg). Tỷ lệ các mạch máu nhỏ được tưới máu tốt có liên quan nghịch đảo với Psl-aCO2 (R2 = 0,80, p < 0,01), cho thấy sự gia tăng tỷ lệ mao mạch được tưới máu tốt song song với sự thay đổi nghịch đảo của áp suất CO2 mô. Tương tự, Nevière và các đồng nghiệp [48] nhận thấy rằng những thay đổi tạo ra bởi dobutamin trong lưu lượng máu vi tuần hoàn dạ dày được phản ánh tốt bởi những thay đổi trong sự khác biệt CO2 từ niêm mạc đến động mạch (PGtis-aCO2), do đó hỗ trợ vai trò hàng đầu của lưu lượng máu vi tuần hoàn trong việc tích lũy CO2 mô dạ dày.
Các quan sát gần đây cũng cho thấy mối quan hệ chặt chẽ giữa thay đổi lưu lượng máu vi tuần hoàn và Pv-aCO2 trong giai đoạn đầu của shock nhiễm trùng [11]. Đặc biệt, sự không đồng nhất của lưu lượng máu vi tuần hoàn và giảm mật độ mao mạch chức năng có liên quan tốt với sự gia tăng lũy tiến của Pv-aCO2. Điều thú vị là, sự thay đổi trong cung lượng tim không tương quan tốt với sự thay đổi các thông số lưu lượng máu vi tuần hoàn, mặc dù phải thừa nhận rằng giá trị Pv-aCO2 cao hơn thường được quan sát thấy ở các giá trị cung lượng tim thấp hơn.
Trong quá trình thiếu oxy mô, tổng VCO2 giảm mặc dù có một CO2 kỵ khí hình thành. Tuy nhiên, CCO2 tĩnh mạch sẽ tăng lên khi lưu lượng máu vĩ mô giảm hoặc lưu lượng máu vi tuần hoàn trở nên không đồng nhất. Trên thực tế, khi xem xét VCO2 không đổi, CO2 tĩnh mạch sẽ tăng ngay cả ở các giá trị cung lượng tim bình thường, khi lưu lượng máu vi tuần hoàn trở nên không đồng nhất và mật độ mao mạch giảm (Hình 16.3). Theo cách này, việc theo dõi các khoảng trống PCO2 có thể cung cấp thông tin quan trọng về sự thay đổi lưu lượng máu vi tuần hoàn ngay cả ở những bệnh nhân có cung lượng tim và các thông số có nguồn gốc oxy bình thường.
Giá trị lâm sàng của Pv-aCO2
Các thay đổi Pv-aCO2 phản ánh các biến đổi lưu lượng máu vĩ mô trong các điều kiện không viêm nhiễm bất thường như ngừng tim, shock giảm thể tích hoặc xuất huyết và chèn ép tim [24-28]. Tuy nhiên, trong quá trình shock nhiễm trùng, Pv-aCO2 có thể bị ảnh hưởng bởi sự phân phối lưu lượng máu vi tuần hoàn, do đó mối quan hệ giữa cung lượng tim và Pv-aCO2 không được quan sát nhất quán trong các nghiên cứu lâm sàng và thực nghiệm.
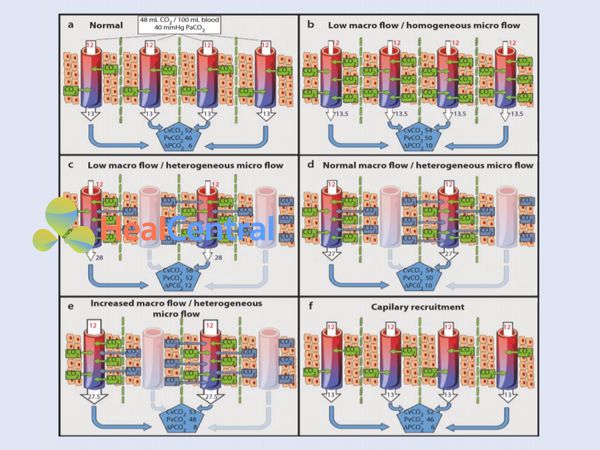
Hình 16.3 Các biến thể lưu lượng máu vĩ mô và vi tuần hoàn và ảnh hưởng của nó đến Pv-aCO2. Bảng a điều kiện bình thường của lưu lượng máu vĩ mô và vi mô: 48 mL CO2/dL (hoặc PvCO2 40 mmHg) được tiến hành trong suốt bốn mao mạch với dòng chảy đối lưu liên tục bình thường (mũi tên trắng mao mạch). Máu đầu tĩnh mạch sẽ được nạp CO2 tạo ra từ trao đổi chất hiếu khí dẫn đến CvCO2 là 52 mL/dL (hoặc PvCO2 46 mmHg) tạo ra Pv-aCO2 là 6 mmHg. Bảng b Cung lượng tim thấp với lưu lượng máu vi mạch đồng nhất. Thời gian vận chuyển chậm của máu mao mạch (mũi tên trắng mỏng trong mao mạch) sẽ dẫn đến tải CO2 cao hơn ở máu ra đầu tĩnh mạch ngay cả khi chuyển hóa hiếu khí được duy trì (hiện tượng đình trệ). Máu tĩnh mạch sẽ được nạp thêm CO2 hiếu khí dẫn đến CvCO2 là 54 mL/dL (hoặc PvCO2 50 mmHg) và Pv-aCO2 là 10 mmHg. Bảng c, Cung lượng tim thấp và lưu lượng máu vi mạch không đồng nhất. Các mao mạch thông suốt sẽ được nạp một lượng CO2 hiếu khí cao hơn từ các nhóm tế bào lân cận do ứ đọng mạch máu (mũi tên trắng mỏng trong các mao mạch mở thể hiện lưu lượng đối lưu thấp). Hơn nữa, máu đi qua tĩnh mạch cũng sẽ được nạp thêm CO2 từ các nhóm tế bào ở xa. Lượng CO2 bổ sung này sẽ là một phần của sản phẩm từ quá trình chuyển hóa hiếu khí giảm đi, và hầu hết sẽ tiến hành từ quá trình tạo CO2 yếm khí do hệ thống đệm giải phóng H+ (xem bài để biết chi tiết). Do đó, máu tĩnh mạch sẽ được nạp thêm CO2 hiếu khí và kỵ khí dẫn đến CvCO2 là 56 mL / dL (hoặc PvCO2 52 mmHg) và Pv-aCO2 là 12 mmHg. Bảng d, Cung lượng tim bình thường và lưu lượng máu vi mạch không đồng nhất. CO2 kỵ khí sẽ được tạo ra trong máu dưới dạng sản phẩm hệ đệm giải phóng H+ (tăng do hạn chế O2 thứ phát do phân phối sai lưu lượng máu – xem bài để biết chi tiết). Mặc dù cung lượng tim bình thường rõ ràng, nhưng điều này là không đủ để “rửa trôi” lượng CO2 dư thừa. Do đó, máu tĩnh mạch sẽ được nạp thêm CO2 hiếu khí và kỵ khí dẫn đến CvCO2 là 54 mL/dL (hoặc PvCO2 50 mmHg), và Pv-aCO2 sẽ vẫn ở mức cao (10 mmHg). Bảng e, Tăng cung lượng tim nhưng lưu lượng máu vi tuần hoàn không đồng nhất. Lưu lượng vĩ mô tăng sẽ rửa cả CO2 hiếu khí và kỵ khí được tạo ra trong các mô (mũi tên trắng dày trong suốt mao mạch mở thể hiện lưu lượng đối lưu tăng). Tuy nhiên, khi lưu lượng máu vi mạch không đồng nhất nghiêm trọng, CO2 kỵ khí sẽ tăng trong máu tĩnh mạch mặc dù giá trị cung lượng tim cao rõ ràng. Do đó, máu tĩnh mạch sẽ được nạp thêm CO2 hiếu khí và kỵ khí dẫn đến CvCO2 là 53 mL/dL (hoặc PvCO2 48 mmHg) và Pv-aCO2 là 8 mmHg. Bảng f trường hợp huy động được mao mạch. Các biện pháp can thiệp cải thiện phân phối lưu lượng máu vi mạch dẫn đến bình thường hóa Pv- aCO2 mà thậm chí cung lượng tim giảm rõ rệt từ mức “cao” đến mức “bình thường”. Lưu ý: các đường chấm thẳng đứng màu xanh lá cây đại diện cho các giới hạn của các khu vực hình trụ lý thuyết phụ thuộc vào mỗi mạch máu mao mạch.
Những quan sát sớm trong shock nhiễm trùng đã chứng minh rằng những bệnh nhân có Pv-aCO2 > 6 mmHg cho thấy giá trị cung lượng tim thấp hơn so với những người có Pv-aCO2 ≤ 6 mmHg [36]. Điều thú vị là, những người đáp ứng dương tính với dịch truyền biểu hiện giảm đồng thời Pv-aCO2, mặc dù đồng thuận về mặt toán học giữa Pv-aCO2 và thay đổi cung lượng tim thực sự kém (r = 0,42 hoặc R2 = 0,18, p < 0,001). Tương tự, mối quan hệ nghịch đảo (mặc dù yếu về mặt toán học) giữa cung lượng tim và Pv-aCO2 (r = 0,41 hoặc R2 = 0,17, p < 0,001) đã được báo cáo bởi Bakker et al. [35], và mặc dù cung lượng tim và DO2 thấp hơn ở những bệnh nhân có Pv-aCO2 > 6 mmHg, VO2 giống hệt nhau đã được quan sát thấy ở cả hai nhóm do thay đổi ERO2 thích nghi.
Pv-aCO2 > 6.0 mmHg ở bệnh nhân bị shock nhiễm trùng đạt ScvO2 > 70% sau khi hồi sức ban đầu cũng có liên quan đến rối loạn chức năng đa cơ quan nặng hơn [15]. Tương tự, Ospina-Tascón et al cho thấy Pv- aCO2 dai dẵng trong giai đoạn sớm của hồi sức shock nhiễm trùng liên quan đến hội chứng suy đa cơ quan nhiều hơn và kết cuộc xấu hơn ở ngày thứ 28. Tăng Pv-aCO2 đã được cho có liên quan đến các biến cố kết cuộc lâm sàng xấu ngay khi ScvO2 đạt được mục tiêu. Hơn nữa, nồng độ Lactate cao hơn và phục hồi Lactate chậm hơn đã được quan sát thấy ở những bệnh nhân có giá trị Pv-aCO2 cao liên tục trong 6 giờ đầu hồi sức. Quan trọng là, khoảng trống PCO2 thu được từ các mẫu máu tĩnh mạch trung tâm và tĩnh mạch hỗn hợp thể hiện sự thỏa thuận tốt. Tuy nhiên, như được đề xuất trong một nghiên cứu gần đây ở bệnh nhân shock nhiễm trùng [37], không có thỏa thuận nào được quan sát giữa Pv-aCO2 và cung lượng tim.
Điều thú vị là, mặc dù Pv-aCO2 có liên quan đến kết quả bất lợi trong shock nhiễm trùng [14,15,37,49,50] và các thủ thuật phẫu thuật có nguy cơ cao [51], giá trị tiên đoán của nó trong phẫu thuật tim vẫn còn gây tranh cãi [52, 53].
Do đó, Pv-aCO2 cao có thể nhận dạng bệnh nhân nhiễm trùng huyết vẫn được hồi sức không đầy đủ mặc dù đạt được các mục tiêu chuyển hóa oxy, củng cố quan niệm về Pv-aCO2 như một dấu hiệu của tưới máu toàn thân do khả năng phát hiện sự thay đổi lưu lượng máu. Tuy nhiên, Pv-aCO2 bình thường có thể không phát hiện ra sự thiếu oxy của mô vì giá trị cung lượng tim tăng cao có thể ngăn ngừa tăng CO2 tĩnh mạch bằng cách đơn giản là rửa trôi mô.
Tỷ lệ CO2 tĩnh-động mạch và O2 động-tĩnh mạch (Cv- aCO2/Ca-vO2 Ratio)
Lý do về mặt sinh lý học
Theo phương trình Fick, mức tiêu thụ oxy (VO2) và sản xuất CO2 (VCO2) tỷ lệ thuận với cung lượng tim và sự khác biệt về hàm lượng từ động mạch đến tĩnh mạch và tĩnh mạch so với động mạch. Trong điều kiện trạng thái ổn định hiếu khí, VCO2 tiệm cận VO2, theo đó sự khác biệt về hàm lượng CO2 từ tĩnh mạch đến động mạch (Cv-aCO2) gần bằng với chênh lệch hàm lượng O2 từ động mạch đến hỗn hợp (Ca-vO2). Theo đó, VCO2 không được vượt quá mức khả dụng của O2 và do đó, tỷ lệ VCO2/VO2 (tức là, chỉ số hô hấp (RQ)) không được > 1.0 trong điều kiện nghỉ ngơi hiếu khí như vậy. Tỷ lệ Cv-aCO2/Ca-vO2 có thể là sự thay thế của tỷ lệ VCO2/VO2 hoặc RQ, và ở một mức độ nào đó, nó phải độc lập với các biến đổi dòng chảy, theo phương trình Fick, cung lượng tim có trong cả hai thành phần tử số và mẫu số. Một phân tích gần đây từ một mô hình shock mất máu tiến triển thực nghiệm cho thấy tỷ lệ Pv-aCO2/Ca- vO2 là đại diện kém cho chuyển hóa kỵ khí trong quá trình pha loãng máu [54]. Tuy nhiên, các tác giả khác đã quan sát thấy sự gia tăng đồng thời tỷ lệ Cv-aCO2/Ca-vO2, chỉ số hô hấp (được đo bằng phép đo nhiệt lượng – calorimetry gián tiếp) và nồng độ lactate trong suy tuần hoàn ở bệnh nhân thở máy [55], do đó củng cố ý tưởng về mối liên kết giữa chuyển hóa kỵ khí và tăng tỷ lệ Cv-aCO2/Ca-vO2.
Sự phong tỏa thực nghiệm việc sử dụng O2 của ty thể dẫn đến việc giảm không đối xứng trong VCO2 và VO2 khi tăng RQ. Sự sụt giảm VCO2/VO2 không đối xứng này có thể được giải thích bằng việc tăng sản xuất CO2 yếm khí có nguồn gốc từ việc hệ đệm quá nhiều proton (chủ yếu được phân phối trong quá trình thủy phân ATP) không được xoay vòng trong quá trình phosphoryl hóa oxy hóa do kết quả của sự hạn chế oxy hóa bị hạn chế nghiêm trọng (Hình 16.4 ). Tương tự, trong điều kiện nhu cầu trao đổi chất quá mức (chẳng hạn như trong quá trình tập luyện gắng sức), tổng VCO2 có thể vượt quá mức tăng thích nghi VO2, một khi đạt được ngưỡng yếm khí [56]. Mặt khác, trong shock tuần hoàn, VO2 giảm toàn thân phải đi kèm với giảm VCO2 hiếu khí. Tuy nhiên, các mô hình shock thực nghiệm cũng chứng minh rằng VCO2 có thể giảm nhẹ hơn so với mức giảm VO2 [28, 57], với sự gia tăng tiếp theo về tỷ lệ VCO2/VO2. Điều thú vị là tỷ lệ VCO2/VO2 này trở lại bình thường sau khi đảo ngược shock. Những điều đã nói ở trên cho thấy tỷ lệ Cv-aCO2/Ca- vO2 có thể xác định sự hiện diện của chuyển hóa kỵ khí.
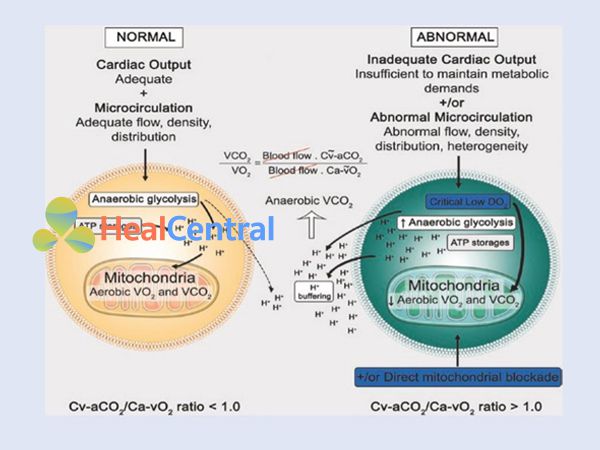
Hình 16.4 Tỷ lệ giữa CO2 tĩnh mạch-động mạch và O2 động mạch-tĩnh mạch (tỷ lệ Cv- aCO2/Ca-vO2). Bình thường hiếu khí (bên trái) và điều kiện yếm khí (bên phải). Cung lượng tim đầy đủ và phân phối lưu lượng máu vi tuần hoàn bình thường thường bảo tồn hô hấp tế bào và tỷ lệ Cv-aCO2/Ca-vO2 vẫn ≤ 1.0. Ngược lại, cung lượng tim không đủ, phân phối sai lệch lưu lượng máu vi tuần hoàn và “phong tỏa” ty thể trực tiếp làm giảm VO2 với VCO2 hiếu khí sau đó. Chuyển hóa tế bào chuyển sang quá trình glycolysis kỵ khí và H+ được giải phóng trong quá trình thủy phân ATP không được tái sử dụng trong quá trình phosphoryl oxy hóa. Do đó, dư H+ không xoay vòng được đệm bởi HCO3 − tạo ra H2CO3, phân tách thành CO2 và H2O. CO2 kỵ khí này sẽ làm tăng tỷ lệ Cv-aCO2/Ca-vO2.
Tỷ lệ Cv-aCO2/Ca-vO2 các ý nghĩa lâm sàng
Theo truyền thống, tăng lactate máu đã được công nhận là một dấu hiệu của sự trao đổi chất kỵ khí thứ phát do việc cung cấp oxy không đủ cho các tế bào. Tuy nhiên, nồng độ lactate huyết tương có thể tăng do các nguyên nhân khác ngoài tình trạng thiếu oxy mô [58]. Trên thực tế, mức độ lactate cao có thể thường xuyên là kết quả của tình trạng tăng hoạt động ly giải đường, chuyển hóa pyruvate bất thường hoặc thay đổi độ thanh thải chuyển hóa lactate [59-61], cản trở sự giải thích của nó trong giai đoạn hồi sức và sau hồi sức. Sử dụng áp suất riêng phần CO2 thay vì hàm lượng CO2, MekontsoDessap et al. [12] cho thấy một thỏa thuận tốt giữa tỷ lệ Pv-aCO2/Ca-vO2 (thay thế cho tỷ lệ Cv-aCO2/Ca- vO2) và mức độ lactate ≥ 2.0 mmol/L (chấp nhận nó là chỉ số chuyển hóa kỵ khí). Tuy nhiên, quan trọng hơn là một thỏa thuận đơn giản, tỷ lệ Cv- aCO2/Ca-vO2 có thể cung cấp thêm thông tin bổ sung mà vốn dĩ được cung cấp bởi nồng độ lactate. Trong một nghiên cứu gần đây, Ospina- Tascón et al. [13] đã chứng minh rằng tăng lactaet kéo dài kết hợp với tỷ lệ Cv-aCO2/Ca-vO2 cao có liên quan đến rối loạn chức năng cơ quan nghiêm trọng hơn và kết quả lâm sàng tồi tệ hơn trong shock nhiễm trùng so với những bệnh nhân có mức độ lactate bình thường và tỷ lệ Cv-aCO2/Ca-vO2 ≤ 1,0. Điều thú vị là, bệnh nhân đạt được mức độ lactate < 2,0 mmol/L nhưng với tỷ lệ Cv-aCO2/Ca-vO2 tăng cao liên tục có kết quả lâm sàng tương tự so với những người có tăng lactate kéo dài và tỷ lệ Cv-aCO2/Ca-vO2 bình thường. Tuy nhiên, liệu tỷ lệ Cv-aCO2/Ca-vO2 tăng có thể đi trước sự gia tăng nồng độ lactate trong shock nhiễm trùng hay không nên được xác nhận trong tương lai.
Các nghiên cứu sau đó đã chứng minh tỷ lệ Cv-aCO2/Ca-vO2 là yếu tố tiên lượng trong shock nhiễm trùng [62, 63], trong khi các nghiên cứu khác cho rằng tỷ lệ Cv-aCO2/Ca-vO2 cao và lactate cao có thể xác định tình trạng phụ thuộc vào tỷ lệ VO2/DO2 [64, 65]. Đồng ý với khái niệm này, các tác giả khác đã chứng minh rằng VO2 tăng sau khi tải dịch truyền chỉ ở những bệnh nhân bị suy tuần hoàn cấp tính và tăng tỷ lệ Pv- aCO2/Ca-vO2 trước khi truyền dịch [64, 65]. Hơn nữa, bằng chứng từ các mô hình shock nhiễm trùng thực nghiệm cho thấy rằng cải thiện phân phối lưu lượng máu vi tuần hoàn có thể đảo ngược quá trình chuyển hóa yếm khí được phản ánh bởi tỷ lệ giảm trong tỷ lệ Cv-aCO2/Ca-vO2 [41].
Tóm lại, tỷ lệ Cv-aCO2/Ca-vO2 hoặc tương đương, tỷ lệ Pv-aCO2/Ca- vO2 (với những hạn chế rõ ràng vì hiệu ứng Haldane), có thể cung cấp thông tin tiên lượng quan trọng và có thể giúp làm rõ nguồn gốc tăng lactate (từ bản chất hiếu khí hoặc kỵ khí) trong giai đoạn đầu của shock. Tỷ lệ Cv-aCO2/Ca-vO2 phản ứng nhanh hơn mức độ lactate với những thay đổi huyết động ngắn hạn, khiến nó trở thành một biến số hấp dẫn cần theo dõi, và mặc dù khó tính toán nhưng việc giải thích nó dễ dàng hơn, với các giá trị > 1.0 có thể gợi ý sự hiện diện của quá trình chuyển hóa kỵ khí đang diễn ra.
Pv-aCO2 và hiệu ứng Haldane
Liên kết CO2 với hemoglobin sẽ thay đổi tùy theo trạng thái oxy hóa hoặc khử oxy của hemoglobin. Hiện tượng này được gọi là hiệu ứng Haldane cho phép nạp CO2 từ các mô vào máu tốt hơn khi oxy di chuyển theo hướng ngược lại, do đó làm tăng khả năng mang CO2 của máu tĩnh mạch. Ngược lại, oxy di chuyển từ phế nang đến máu mao mạch giúp tăng cường không nạp CO2 từ huyết sắc tố, do đó tạo điều kiện cho việc loại bỏ phổi của nó. Do đó, các giá trị bão hòa oxy thấp làm tăng hàm lượng CO2 (CCO2) cho một pCO2 nhất định, theo đó, nhiều CO2 sẽ được liên kết với hemoglobin. Những thay đổi trong chiết xuất oxy mô, pH, VCO2 và nồng độ hemoglobin có thể ảnh hưởng đến Pv-aCO2 mặc dù tưới máu mô được bảo tồn hoặc thậm chí tăng. Ví dụ, sự kết hợp của lưu lượng máu cao và sự gia tăng VCO2 lớn hơn so với sự thay đổi tương ứng trong mức tiêu thụ oxy có thể dẫn đến sự phân ly của các gradient CO2 từ mô đến động mạch hoặc tĩnh mạch đến động mạch giữa các giường mạch máu khác nhau với các giá trị ERO2 nền khác nhau. Trên thực tế, sự gia tăng nghịch lý về sự khác biệt pCO2 từ niêm mạc đến động mạch có thể xảy ra trong quá trình tăng lưu lượng máu do sự thay đổi của bão hòa oxy tĩnh mạch, VCO2 cục bộ hoặc cả hai [66].
Tùy thuộc vào SvO2 nền, hiệu ứng Haldane có thể tăng hoặc giảm Pv-aCO2 để đáp ứng với những thay đổi tương tự trong lưu lượng máu hoặc chuyển hóa [67]. Phải thừa nhận rằng Pv-aCO2/ Ca-vO2 có thể tương đương với tỷ lệ Cv-aCO2/Ca-vO2 khi pCO2, pH và SvO2 gần bình thường, xảy ra thường xuyên. Tuy nhiên, Cv-aCO2 không phải lúc nào cũng được đại diện bởi Pv-aCO2, đặc biệt là trong điều kiện pCO2 và SvO2 thấp. Về vấn đề này, một nghiên cứu gần đây đã chứng minh giá trị tiên lượng của tỷ lệ Cv-aCO2/ Ca-vO2 trong shock nhiễm trùng và độ tin cậy tương đương của nó về áp lực riêng phần, tỷ lệ Pv-aCO2/ Ca-vO2 [13]. Do đó, mặc dù ở các giá trị Pv-aCO2 thấp, ảnh hưởng của hiệu ứng Haldane là không đáng kể, sự phân tán của Cv-aCO2 so với Pv-aCO2 trở nên rộng hơn đáng kể ở các giá trị Pv-aCO2 cao hơn [13].
Sự đơn giản của phép đo Pv-aCO2 làm cho nó trở thành một công cụ hấp dẫn để hướng dẫn hồi sức trong môi trường lâm sàng. Tuy nhiên, Pv-aCO2 là một phép đo phức tạp về sinh lý nên được giải thích theo một số biến số sinh lý.
Giải thích Pv-aCO2 và tỷ lệ Cv-aCO2/Ca-vO2 trong shock nhiễm trùng
Sự khác biệt CO2 từ mô đến động mạch và tĩnh mạch đến động mạch nên được coi là dấu hiệu của tưới máu mô hơn là các chỉ số của tình trạng thiếu oxy mô. Sự đồng hành của mức Pv-aCO2 cao (> 6.0 mmHg) và nồng độ SvO2 thấp thường phản ánh cung lượng tim thấp trong cả điều kiện viêm và không viêm. Tương tự, SvO2 bình thường đi kèm với Pv-aCO2 tăng liên tục cho thấy sự hiện diện của cung lượng tim không đủ để làm sạch CO2 do các mô tạo ra. Ngoài ra, các giá trị Pv-aCO2 cao với các giá trị SvO2 bình thường hoặc thậm chí cao trùng khớp với các rối loạn vi tuần hoàn như giảm mật độ mao mạch chức năng hoặc tăng sự không đồng nhất của lưu lượng máu vi mạch, ít nhất là trong giai đoạn đầu của shock nhiễm trùng [11, 45]. Trong mọi trường hợp, Pv- aCO2 tăng phản ánh lưu lượng máu vĩ mô hoặc vi mô thay đổi độc lập với sự hiện diện của chuyển hóa kỵ khí. Do đó, Pv-aCO2 tăng cao nên khuyến khích các bác sĩ lâm sàng tối ưu hóa cung lượng tim hoặc có thể huy động vi tuần hoàn để cải thiện tưới máu mô, đặc biệt là khi tăng mức độ lactate và có dấu hiệu lâm sàng của giảm tưới máu. Tuy nhiên, những quyết định như vậy cần tính đến bối cảnh lâm sàng và thông tin được cung cấp bởi giám sát “đa phương thức” [68]. Trong điều kiện hiếu khí, Pv-aCO2 cao có nghĩa là lưu lượng máu không đủ ngay cả khi cung lượng tim ở trong phạm vi bình thường. Trong bối cảnh này, những nỗ lực tiếp theo để tăng cung lượng tim nhằm ngăn chặn tình trạng thiếu oxy có thể xảy ra vẫn còn gây tranh cãi và cần được đánh giá trong tương lai.
Trong điều kiện phụ thuộc vào việc cung cấp oxy, sự gia tăng cung lượng tim phải đi kèm với sự gia tăng của VO2 và do đó, bằng cách tăng VCO2 hiếu khí, do đó Pv-aCO2 có thể giảm ở mức độ thấp hơn sau khi can thiệp tích cực như vậy. Do đó, Pv-aCO2 giảm nhẹ không phải lúc nào cũng có nghĩa là can thiệp điều trị không hiệu quả. Do đó, trong trường hợp phụ thuộc vào nguồn cung cấp oxy có thể xảy ra, các biện pháp can thiệp tối ưu hóa cung lượng tim có lẽ nên được duy trì cho đến khi thu được giá trị Pv-aCO2 giảm. Đáng chú ý, hầu hết các biện pháp can thiệp nhằm tăng cung lượng tim sẽ làm tăng VCO2 do các amin gây co mạch và inotropin tích cực làm tăng hiệu ứng sinh nhiệt [69]. Về vấn đề này, Pv- aCO2 có thể được sử dụng như một chỉ số phản ánh mối quan hệ cung cấp VCO2 / tim [29], và do đó, nó có thể giúp điều chỉnh thuốc [70]. Ngược lại, Pv-aCO2 bình thường (<6.0 mmHg) cho thấy rằng cung lượng tim đủ để làm sạch CO2 do các mô tạo ra và cũng cho thấy lưu lượng máu vi tuần hoàn được phân phối đầy đủ. Tuy nhiên, liệu có nên xử lý cung lượng tim hoặc vi tuần hoàn trong điều kiện thiếu oxy rõ ràng với Pv- aCO2 < 6.0 mmHg hay không.
Tỷ lệ Cv-aCO2/ Ca-vO2> 1.0 có thể gợi ý sự hiện diện của chuyển hóa kỵ khí.
Do đó, việc kết hợp nồng độ lactate và tỷ lệ Cv-aCO2/ Ca-vO2 có thể cung cấp thông tin liên quan trong giai đoạn đầu của quá trình hồi sức. Nồng độ tăng lactate đi kèm với tỷ lệ Cv-aCO2/ Ca-vO2 > 1.0 có thể gợi ý chuyển hóa kỵ khí “đang diễn ra”; do đó, các bác sĩ lâm sàng nên được khuyến khích để tối ưu hóa cả các thông số lưu lượng máu vĩ mô và vi mô. Ngược lại, mức độ tăng của lactate đi kèm với tỷ lệ Cv-aCO2/ Ca-vO2 ≤ 1.0 có thể gợi ý sự tích lũy lactate do rối loạn chức năng tế bào khi có sự trao đổi chất hiếu khí. Trong những trường hợp như vậy, các thao tác hồi sức bổ sung nhằm mục đích tăng lưu lượng máu có lẽ không nên được khuyến khích, mặc dù điều này cần được xác nhận trong các thử nghiệm lâm sàng. Với phản ứng nhanh hơn trong các biến số liên quan CO2, tỷ lệ Cv-aCO2/ Ca-vO2 > 1.0 với mức độ lactate bình thường cuối cùng có thể gợi ý sự khởi đầu của quá trình chuyển hóa yếm khí, thậm chí dự đoán sự gia tăng nồng độ lactate. Tuy nhiên, sự phức tạp của tỷ lệ Cv- aCO2/ Ca-vO2 đáng để nghiên cứu và xác nhận thêm trong môi trường lâm sàng.
Kết luận
Sinh lý xác định tăng CO2 tĩnh mạch là phức tạp. Tuy nhiên, Pv-aCO2 trên toàn hệ thống phản ánh sự thay đổi lưu lượng máu ở cả cấp độ vĩ mô và vi mô, hơn là rối loạn oxy tại mô. Trong khi đó, tỷ lệ Cv-aCO2/ Ca- vO2 tăng cao có thể phản ánh quá trình chuyển hóa yếm khí và nó có thể thêm thông tin tiên lượng quan trọng ở bệnh nhân bị shock. Mặc dù các cơ sở sinh lý của việc theo dõi các biến có nguồn gốc CO2 như vậy, tiện ích lâm sàng của nó trong quá trình hồi sức trong shock vẫn được chứng minh trong các nghiên cứu thực nghiệm và lâm sàng trong tương lai.
Thông tin cốt lõi
Pv-aCO2 được xác định bởi sự kết hợp của lưu lượng máu vĩ mô và/hoặc vi mô, tổng sản xuất CO2 (cả hiếu khí và kỵ khí) và mối quan hệ phức tạp giữa áp lực riêng phần CO2 và hàm lượng máu CO2 (hiệu ứng Haldane).
Pv-aCO2 nên được coi là một dấu hiệu của tưới máu mô nhưng không phải là thiếu oxy mô.
Một Pv-aCO2 tăng lên thường gợi ý một cung lượng tim “thấp” và hoặc “không đủ”. Tuy nhiên, trong điều kiện viêm nặng, sự thay đổi mật độ mao mạch chức năng và sự không đồng nhất của lưu lượng máu vi tuần hoàn cũng có thể giải thích cho sự tích tụ CO2 tĩnh mạch.
Pv-aCO2 tăng cao nên khuyến khích các bác sĩ lâm sàng tối ưu hóa cung lượng tim, đặc biệt là khi nồng độ lactate tăng lên và có dấu hiệu lâm sàng của giảm tưới máu.
Trong điều kiện hiếu khí, những nỗ lực tiếp theo để tăng cung lượng tim nhằm ngăn chặn tình trạng thiếu oxy có thể xảy ra khi có Pv- aCO2 cao vẫn còn gây tranh cãi.
Một tỷ lệ khác biệt giữa carbon dioxide tĩnh mạch-động mạch so với chênh lệch hàm lượng oxy động mạch-tĩnh mạch (Cv-aCO2/ Ca-vO2) có thể phản ánh sự hiện diện của chuyển hóa yếm khí. Có một số bằng chứng thực nghiệm cho thấy tỷ lệ Cv-aCO2/ Ca-vO2 cao có thể được đảo ngược bằng các thao tác hồi sức, ít nhất là trong giai đoạn đầu của shock.
Tỷ lệ Cv-aCO2/ Ca-vO2 cao có thể cung cấp thêm thông tin tiên lượng trong shock nhiễm trùng. Liệu tỷ lệ Cv-aCO2/ Ca-vO2 có thể dự đoán sự gia tăng của lactate trong giai đoạn đầu của shock hay không vẫn còn được làm sáng tỏ.
TÀI LIỆU THAM KHẢO
1. Cecconi M, De Backer D, Antonelli M, Beale R, Bakker J, Hofer C, et al. Consensus on circulatory shock and hemodynamic monitoring. Task force of the European Society of Intensive Care Medicine. Intensive Care Med. 2014;40(12):1795–815.
2. Vincent JL, De Backer D. Circulatory shock. N Engl J Med. 2013;369(18):1726–34.
3. Shoemaker WC, Appel PL, Kram HB. Tissue oxygen debt as a determinant of lethal and nonlethal postoperative organ failure. Crit Care Med. 1988;16(11):1117–20.
4. Vallet B. Vascular reactivity and tissue oxygenation. Intensive Care Med. 1998;24(1):3–11.
5. Rivers E, Nguyen B, Havstad S, Ressler J, Muzzin A, Knoblich B, et al. Early goal-directed therapy in the treatment of severe sepsis and septic shock. N Engl J Med. 2001;345(19):1368–77.
6. Bellomo R, Reade MC, Warrillow SJ. The pursuit of a high central venous oxygen saturation in sepsis: growing concerns. Crit Care. 2008;12(2):130.
7. Peake SL, Delaney A, Bailey M, Bellomo R, Cameron PA, Cooper DJ, et al. Goal-directed resuscitation for patients with early septic shock. N Engl J Med. 2014;371(16):1496–506.
8. Mouncey PR, Osborn TM, Power GS, Harrison DA, Sadique MZ, Grieve RD, et al. Trial of early, goal-directed resuscitation for septic shock. N Engl J Med. 2015;372(14):1301–11.
9. Yealy DM, Kellum JA, Huang DT, Barnato AE, Weissfeld LA, Pike F, et al. A randomized trial of protocolbased care for early septic shock. N Engl J Med. 2014;370(18):1683–93.
10. van Beest PA, Hofstra JJ, Schultz MJ, Boerma EC, Spronk PE, Kuiper MA. The incidence of low venous oxygen saturation on admission to the intensive care unit: a multi-center observational study in The Netherlands. Crit Care. 2008;12(2):R33.
11. Ospina-Tascón GA, Umaña M, Bermúdez WF, Bautista-Rincón DF, Valencia JD, Madriñán HJ, et al. Can venous-to-arterial carbon dioxide differences reflect microcirculatory alterations in patients with septic shock? Intensive Care Med. 2016;42(2):211–21.
12. Mekontso-Dessap A, Castelain V, Anguel N, Bahloul M, Schauvliege F, Richard C, et al. Combination of venoarterial PCO2 difference with arteriovenous O2 content difference to detect anaerobic metabolism in patients. Intensive Care Med. 2002;28(3):272–7.
13. Ospina-Tascón GA, Umaña M, Bermúdez W, Bautista-Rincón DF, Hernandez G, Bruhn A, et al. Combination of arterial lactate levels and venous-arterial CO2 to arterial-venous O 2 content difference ratio as markers of resuscitation in patients with septic shock. Intensive Care Med. 2015;41(5):796–805.
14. Ospina-Tascón GA, Bautista-Rincón DF, Umaña M, Tafur JD, Gutiérrez A, García AF, et al. Persistently high venous-to-arterial carbon dioxide differences during early resuscitation are associated with poor outcomes in septic shock. Crit Care. 2013;17(6):R294.
15. Vallée F, Vallet B, Mathe O, Parraguette J, Mari A, Silva S, et al. Central venous-to-arterial carbon dioxide difference: an additional target for goal- directed therapy in septic shock? Intensive Care Med. 2008;34(12):2218–25.
16. Herve P, Simonneau G, Girard P, Cerrina J, Mathieu M, Duroux P. Hypercapnic acidosis induced by nutrition in mechanically ventilated patients: glucose versus fat. Crit Care Med. 1985;13(7):537–40.
17. Marcinek DJ, Kushmerick MJ, Conley KE. Lactic acidosis in vivo: testing the link between lactate generation and H+ accumulation in ischemic mouse muscle. J Appl Physiol (1985). 2010;108(6):1479–86.
18. Randall HM, Cohen JJ. Anaerobic CO2 production by dog kidney in vitro. Am J Phys. 1966;211(2): 493–505.
19. Jensen FB. Comparative analysis of autoxidation of haemoglobin. J Exp Biol. 2001;204(Pt 11):2029–33.
20. McHardy GJ. The relationship between the differences in pressure and content of carbon dioxide in arterial and venous blood. Clin Sci. 1967;32(2):299–309.
21. Cavaliere F, Giovannini I, Chiarla C, Conti G, Pennisi MA, Montini L, et al. Comparison of two methods to assess blood CO2 equilibration curve in mechanically ventilated patients. Respir Physiol Neurobiol. 2005;146(1):77– 83.
22. Lamia B, Monnet X, Teboul JL. Meaning of arterio-venous PCO2 difference in circulatory shock. Minerva Anestesiol. 2006;72(6):597–604.
23. Giovannini I, Chiarla C, Boldrini G, Castagneto M. Calculation of venoarterial CO2 concentration difference. J Appl Physiol (1985). 1993;74(2):959–64.
24. Grundler W, Weil MH, Rackow EC. Arteriovenous carbon dioxide and pH gradients during cardiac arrest. Circulation. 1986;74(5):1071–4.
25. Weil MH, Rackow EC, Trevino R, Grundler W, Falk JL, Griffel MI. Difference in acid-base state between venous and arterial blood during cardiopulmonary resuscitation. N Engl J Med. 1986;315(3):153–6.
26. Zhang H, Vincent JL. Arteriovenous differences in PCO2 and pH are good indicators of critical hypoperfusion. Am Rev Respir Dis. 1993;148(4 Pt 1):867–71.
27. Van der Linden P, Rausin I, Deltell A, Bekrar Y, Gilbart E, Bakker J, et al. Detection of tissue hypoxia by arteriovenous gradient for PCO2 and pH in anesthetized dogs during progressive hemorrhage. Anesth Analg. 1995;80(2):269–75.
28. Groeneveld AB, Vermeij CG, Thijs LG. Arterial and mixed venous blood acid-base balance during hypoperfusion with incremental positive end- expiratory pressure in the pig. Anesth Analg. 1991;73(5): 576–82.
29. Teboul JL, Mercat A, Lenique F, Berton C, Richard C. Value of the venous- arterial PCO2 gradient to reflect the oxygen supply to demand in humans: effects of dobutamine. Crit Care Med. 1998;26(6):1007–10.
30. Schlichtig R, Bowles SA. Distinguishing between aerobic and anaerobic appearance of dissolved CO2 in intestine during low flow. J Appl Physiol (1985). 1994;76(6):2443–51.
31. Vallet B, Tavernier B, Lund N. Assessment of tissue oxygenation in the critically III. In: Vincent J-L, editor. Yearbook of intensive care and emergency medicine. Berlin/Heidelberg: Springer Berlin Heidelberg; 2000. p. 715–25.
32. Vallet B, Teboul JL, Cain S, Curtis S. Venoarterial CO(2) difference during regional ischemic or hypoxic hypoxia. J Appl Physiol (1985). 2000;89(4):1317–21.
33. Nevière R, Chagnon JL, Teboul JL, Vallet B, Wattel F. Small intestine intramucosal PCO(2) and microvascular blood flow during hypoxic and ischemic hypoxia. Crit Care Med. 2002;30(2):379–84.
34. Dubin A, Estenssoro E, Murias G, Pozo MO, Sottile JP, Barán M, et al. Intramucosal-arterial Pco2 gradient does not reflect intestinal dysoxia in anemic hypoxia. J Trauma. 2004;57(6):1211–7.
35. Bakker J, Vincent JL, Gris P, Leon M, Coffernils M, Kahn RJ. Veno-arterial carbon dioxide gradient in human septic shock. Chest. 1992;101(2):509–15.
36. Mecher CE, Rackow EC, Astiz ME, Weil MH. Venous hypercarbia associated with severe sepsis and systemic hypoperfusion. Crit Care Med. 1990;18(6):585–9.
37. van Beest PA, Lont MC, Holman ND, Loef B, Kuiper MA, Boerma EC. Central venous-arterial pCO2 difference as a tool in resuscitation of septic patients. Intensive Care Med. 2013;39(6):1034–9.
38. De Backer D, Creteur J, Preiser JC, Dubois MJ, Vincent JL. Microvascular blood flow is altered in patients with sepsis. Am J Respir Crit Care Med. 2002;166(1):98–104.
39. De Backer D, Ospina-Tascon G, Salgado D, Favory R, Creteur J, Vincent JL. Monitoring the microcirculation in the critically ill patient: current methods and future approaches. Intensive Care Med. 2010;36(11):1813–25.
40. Zuurbier CJ, van Iterson M, Ince C. Functional heterogeneity of oxygen supply-consumption ratio in the heart. Cardiovasc Res. 1999;44(3):488–97.
41. Stein JC, Ellis CG, Ellsworth ML. Relationship between capillary and systemic venous PO2 during nonhypoxic and hypoxic ventilation. Am J Phys. 1993;265(2 Pt 2):H537–42.
42. Goldman D, Bateman RM, Ellis CG. Effect of decreased O2 supply on skeletal muscle oxygenation and O2 consumption during sepsis: role of heterogeneous capillary spacing and blood flow. Am J Physiol Heart Circ Physiol. 2006;290(6):H2277–85.
43. Ospina-Tascón GA, García Marin AF, Echeverri GJ, Bermudez WF, Madriñán-Navia H, Valencia JD, et al. Effects of dobutamine on intestinal microvascular blood flow heterogeneity and O2 extraction during septic shock. J Appl Physiol (1985). 2017;122(6):1406–17.
44. Humer MF, Phang PT, Friesen BP, Allard MF, Goddard CM, Walley KR. Heterogeneity of gut capillary transit times and impaired gut oxygen extraction in endotoxemic pigs. J Appl Physiol (1985). 1996;81(2): 895–904.
45. Sakr Y, Dubois MJ, De Backer D, Creteur J, Vincent JL. Persistent microcirculatory alterations are associated with organ failure and death in patients with septic shock. Crit Care Med. 2004;32(9):1825–31.
46. De Backer D, Donadello K, Sakr Y, Ospina-Tascon G, Salgado D, Scolletta S, et al. Microcirculatory alterations in patients with severe sepsis: impact of time of assessment and relationship with outcome. Crit Care Med. 2013;41(3):791–9.
47. Creteur J, De Backer D, Sakr Y, Koch M, Vincent JL. Sublingual capnometry tracks microcirculatory changes in septic patients. Intensive Care Med. 2006;32(4):516–23.
48. Nevière R, Mathieu D, Chagnon JL, Lebleu N, Wattel F. The contrasting effects of dobutamine and dopamine on gastric mucosal perfusion in septic patients. Am J Respir Crit Care Med. 1996;154(6 Pt 1):1684–8.
49. Mallat J, Pepy F, Lemyze M, Gasan G, Vangrunderbeeck N, Tronchon L, et al. Central venous-to-arterial carbon dioxide partial pressure difference in early resuscitation from septic shock: a prospective observational study. Eur J Anaesthesiol. 2014;31(7):371–80.
50. Du W, Liu DW, Wang XT, Long Y, Chai WZ, Zhou X, et al. Combining central venous-to-arterial partial pressure of carbon dioxide difference and central venous oxygen saturation to guide resuscitation in septic shock. J Crit Care. 2013;28(6):1110.e1–5.
51. Robin E, Futier E, Pires O, Fleyfel M, Tavernier B, Lebuffe G, et al. Central venous-to-arterial carbon dioxide difference as a prognostic tool in high- risk surgical patients. Crit Care. 2015;19:227.
52. Guinot PG, Badoux L, Bernard E, Abou-Arab O, Lorne E, Dupont H. Central venous-to-arterial carbon dioxide partial pressure difference in patients undergoing cardiac surgery is not related to postoperative outcomes. J Cardiothorac Vasc Anesth. 2017;31(4):1190–6.
53. Morel J, Grand N, Axiotis G, Bouchet JB, Faure M, Auboyer C, et al. High veno-arterial carbon dioxide gradient is not predictive of worst outcome after an elective cardiac surgery: a retrospective cohort study. J Clin Monit Comput. 2016;30(6):783–9.
54. Dubin A, Ferrara G, Kanoore Edul VS, Martins E, Canales HS, Canullán C, et al. Venoarterial PCO2-to-arteriovenous oxygen content difference ratio is a poor surrogate for anaerobic metabolism in hemodilution: an experimental study. Ann Intensive Care. 2017;7(1):65.
55. Danin PE, Bendjelid K. The venous-arterial CO2 to arterial-venous O2 content difference ratio: easy to monitor? J Crit Care. 2016;35:217–8.
56. Wasserman K, Beaver WL, Whipp BJ. Gas exchange theory and the lactic acidosis (anaerobic) threshold. Circulation. 1990;81(1 Suppl):II14–30. 57. Cohen IL, Sheikh FM, Perkins RJ, Feustel PJ, Foster ED. Effect of hemorrhagic shock and reperfusion on the respiratory quotient in swine. Crit Care Med. 1995;23(3):545–52.
58. Rimachi R, Bruzzi de Carvahlo F, Orellano-Jimenez C, Cotton F, Vincent JL, De Backer D. Lactate/pyruvate ratio as a marker of tissue hypoxia in circulatory and septic shock. Anaesth Intensive Care. 2012;40(3): 427–32.
59. Gore DC, Jahoor F, Hibbert JM, DeMaria EJ. Lactic acidosis during sepsis is related to increased pyruvate production, not deficits in tissue oxygen availability. Ann Surg. 1996;224(1):97–102.
60. Levraut J, Ciebiera JP, Chave S, Rabary O, Jambou P, Carles M, et al. Mild hyperlactatemia in stable septic patients is due to impaired lactate clearance rather than overproduction. Am J Respir Crit Care Med. 1998;157(4 Pt 1):1021–6.
61. Tapia P, Soto D, Bruhn A, Alegría L, Jarufe N, Luengo C, et al. Impairment of exogenous lactate clearance in experimental hyperdynamic septic shock is not related to total liver hypoperfusion. Crit Care. 2015;19:188.
62. He HW, Liu DW, Long Y, Wang XT. High central venous-to-arterial CO2 difference/arterial-central venous O2 difference ratio is associated with poor lactate clearance in septic patients after resuscitation. J Crit Care. 2016;31(1):76–81.
63. Mesquida J, Saludes P, Gruartmoner G, Espinal C, Torrents E, Baigorri F, et al. Central venous-to-arterial carbon dioxide difference combined with arterial-to-venous oxygen content difference is associated with lactate evolution in the hemodynamic resuscitation process in early septic shock. Crit Care. 2015;19:126.
64. Monnet X, Julien F, Ait-Hamou N, Lequoy M, Gosset C, Jozwiak M, et al. Lactate and venoarterial carbon dioxide difference/arterial-venous oxygen difference ratio, but not central venous oxygen saturation, predict increase in oxygen consumption in fluid responders. Crit Care Med. 2013;41(6):1412–20.
65. Mallat J, Lemyze M, Meddour M, Pepy F, Gasan G, Barrailler S, et al. Ratios of central venous-to-arterial carbon dioxide content or tension to arteriovenous oxygen content are better markers of global anaerobic metabolism than lactate in septic shock patients. Ann Intensive Care. 2016;6(1):10. 66. Jakob SM, Kosonen P, Ruokonen E, Parviainen I, Takala J. The Haldane effect – an alternative explanation for increasing gastric mucosal PCO2 gradients? Br J Anaesth. 1999;83(5):740–6.
67. Hurley R, Mythen MG. The Haldane effect – an explanation for increasing gastric mucosal PCO2 gradients? Br J Anaesth. 2000;85(1):167–9.
68. Alegría L, Vera M, Dreyse J, Castro R, Carpio D, Henriquez C, et al. A hypoperfusion context may aid to interpret hyperlactatemia in sepsis-3 septic shock patients: a proof-of-concept study. Ann Intensive Care. 2017;7(1):29.
69. Chioléro R, Flatt JP, Revelly JP, Jéquier E. Effects of catecholamines on oxygen consumption and oxygen delivery in critically ill patients. Chest. 1991;100(6):1676–84.
70. Teboul JL, Graini L, Boujdaria R, Berton C, Richard C. Cardiac index vs oxygen-derived parameters for rational use of dobutamine in patients with congestive heart failure. Chest. 1993;103(1):81–5.
Đăng tải bởi: Heal Central (Health Education Assets Library)




 và (Cv-aCO2/Ca-vO2) trong trạng thái shock){kind=link}