Kháng sinh điều trị lao
Giới thiệu chung
Lịch sử ra đời
Lao là một bệnh nhiễm trùng do vi khuẩn lao Mycobacterium spp. gây ra. Trong đó, loài gây bệnh chủ yếu ở người là M. tuberculosis. Trước đây, lao được liệt vào hàng các bệnh “tứ chứng nan y”, tuy nhiên hiện nay, cùng với sự phát triển của y học hiện đại, lao đã trở thành một bệnh hoàn toàn có thể chữa khỏi nếu được phát hiện sớm và điều trị kịp thời.
Trước đây, khi chưa thể điều trị được bệnh lao, người ta cũng đã biết cách cô lập những bệnh nhân mắc bệnh để hạn chế bệnh có thể truyền từ người bệnh sang người lành, cho họ dinh dưỡng, nghỉ ngơi đầy đủ và tiếp xúc với ánh nắng mặt trời hàng ngày.
Chỉ cho đến tận năm 1882, Robert Koch mới phát hiện ra nguyên nhân gây bệnh, đó là trực khuẩn lao. Sau này, loại vi khuẩn này còn được gọi là trực khuẩn Koch và bản thân ông cũng được trao giải thưởng Nobel về Sinh lý và Y khoa năm 1905. Vài chục năm sau đó, với việc phát hiện ra hoạt tính kháng khuẩn của Penicillin bởi Alexander Fleming, các nhà khoa học đã bắt đầu tập trung đánh giá hiệu lực của các hợp chất hóa học và tự nhiên trong việc điều trị bệnh lao.
Bằng chứng thực nghiệm đầu tiên về hiệu lực tiềm năng của các loại thuốc chống lao mới đã thu được vào năm 1940, khi mà một Sulfonamide có tên gọi là Promin, một dẫn xuất của Dapsone, được sử dụng trên một mẫu chuột lang. Tuy nhiên, bản thân Promin sau này đã không bao giờ được phê duyệt để sử dụng trên người.
Vài năm sau đó, Streptomycin, một kháng sinh nhóm Aminoglycoside được phân lập tự nhiên từ chủng xạ khuẩn Streptomyces griseus, đã cho thấy hiệu quả của nó trên trực khuẩn lao ở cả mô hình động vật cũng như trên người. Năm 1944, Schatz và Waksman đã tuyên bố rằng có thể kê đơn Streptomycin cho những bệnh nhân mắc bệnh lao. Năm 1946, Đơn vị Lao của Hội đồng Nghiên cứu Y khoa Vương quốc Anh đã cho thấy hiệu lực của thuốc trong ngắn hạn (sáu tháng) làm giảm tỷ lệ tử vong từ 27% xuống 7%. Tuy nhiên, sau năm năm, không có sự khác biệt giữa những người sử dụng hoặc không sử dụng Streptomycin. Đây là hậu quả của tình trạng đề kháng kháng sinh.
Bốn năm sau sự ra đời của Streptomycin, một kháng sinh chống lao tổng hợp đầu tiên đã ra đời, đó là Para-aminosalicylic acid (PAS). Nhận thấy hiệu quả đơn trị liệu không đem lại hiệu quả cao, năm 1952, lần đầu tiên phác đồ điều trị lao đa hóa trị sử dụng ba thuốc là Streptomycin, PAS và Isoniazid đã được đề xuất. “Phương pháp Edinburgh” của Sir John Crofton mô tả việc kê đơn ít nhất hai loại thuốc đã cho thấy hiệu lực của phác đồ đa hóa trị.
Năm 1954, Pyrazinamide được khám phá. Tiếp theo sau đó là Ethambutol năm 1961 và Rifampicin năm 1963. Vào thời điểm đó, thời gian điều trị thường kéo dài từ một đến hai năm. Năm 1970, các phác đồ chứa Rifampicin đã cho thấy hiệu quả điều trị tốt trong 9 tháng. Năm 1974, liều thấp Rifampicin và Pyrazinamide đã cho thấy hiệu quả điều trị trong 6 tháng.
Năm 2012, Bedaquiline đã được phê duyệt bởi Cơ quan Quản lý Thực phẩm và Dược phẩm (FDA) Hoa Kỳ. Sau đó một năm, Delamanid đã được Cơ quan Quản lý Châu Âu phê duyệt. Pretomanid là thuốc chống lao mới nhất được phê duyệt hiện nay (2019). Các thuốc chống lao thế hệ mới này được phát triển để điều trị lao đa kháng thuốc.
Dưới đây là danh sách liệt kê các thuốc chống lao đang được sử dụng hiện nay:
- Isoniazid.
- Rifampicin.
- Ethambutol.
- Pyrazinamide.
- Streptomycin.
- Kanamycin.
- Amikacin.
- Capreomycin.
- Ofloxacin.
- Ciprofloxacin.
- Levofloxacin.
- Moxifloxacin.
- Gatifloxacin.
- Ethionamide.
- Prothionamide.
- Para-aminosalicylic acid.
- Cycloserine.
- Clofazimine.
- Linezolid.
- Amoxicillin/Clavulanate.
- Imipenem.
- Clarithromycin.
- Terizidone.
- Thiacetazone.
- Thioridazine.
- Bedaquiline.
- Delamanid.
- Pretomanid.
Các kháng sinh Streptomycin, Kanamycin và Amikacin thuộc nhóm kháng sinh Aminoside (hay Aminoglycoside); Ofloxacin, Ciprofloxacin, Levofloxacin, Moxifloxacin và Gatifloxacin thuộc nhóm kháng sinh Quinolone (cụ thể hơn là các Fluoroquinolone); Linezolid thuộc nhóm kháng sinh Oxazolidinone; Amoxicillin/Clavulanate thuộc nhóm kháng sinh Penicillin (một phân nhóm nhỏ hơn của nhóm kháng sinh β-lactam) và Clarithromycin thuộc nhóm kháng sinh Macrolide. Các kháng sinh này đã được giới thiệu trong các bài viết chuyên biệt về từng nhóm kháng sinh và sẽ ít được nhắc lại trong bài viết này.
Các thuốc điều trị lao được Tổ chức Y tế Thế giới (WHO) phân loại thành năm nhóm, dựa trên một số tiêu chí, trong đó có hiệu lực cũng như các tính chất hóa học đặc trưng. Các thuốc thường được kê đơn cho lao nhạy cảm với thuốc nằm trong nhóm đầu tiên, trong khi các thuốc có hiệu lực không rõ ràng được xếp vào nhóm thứ năm. Nhóm đầu tiên gồm có bốn thuốc: Isoniazid, Rifampicin, Pyrazinamide và Ethambutol. Nhóm thứ hai gồm các thuốc kháng sinh nhóm Aminoside và Capreomycin. Nhóm thứ ba gồm các thuốc kháng sinh nhóm Quinolone. Nhóm thứ tư gồm các thuốc Cycloserine, Ethionamide, Prothionamide, Para-aminosalicylic acid, Terizidone và Thiacetazone. Cuối cùng, nhóm thứ năm bao gồm các thuốc còn lại: Amoxicillin/Clavulanate, Clarithromycin, Clofazimine, Imipenem và Linezolid. Bedaquiline, Delamanid và Pretomanid là các thuốc chống lao mới, dùng để dự trữ cho các trường hợp lao đa kháng thuốc, không đáp ứng với các phác đồ điều trị trước đó.
Cấu trúc hóa học
Các thuốc chống lao có cấu trúc hóa học đa dạng. Nhiều thuốc có cấu trúc rất đơn giản và có thể tổng hợp hóa học cực kỳ dễ dàng.
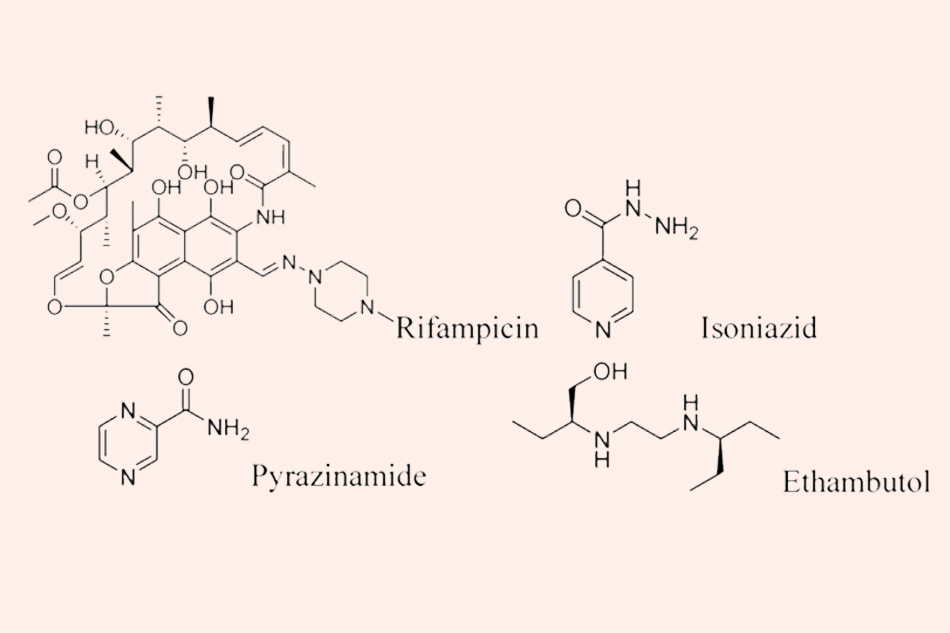
Bốn thuốc sử dụng đầu tay trong điều trị lao hiện nay là Rifampicin, Isoniazid, Pyrazinamide và Ethambutol. Trong đó, Isoniazid, Pyrazinamide và Ethambutol có thể được sản xuất hoàn toàn bằng phương pháp tổng hợp hóa học, còn Rifampicin là một kháng sinh bán tổng hợp từ Rifamycin, chiết xuất từ môi trường nuôi cấy Amycolatopsis rifamycinica, trước đây có tên gọi là Streptomyces mediterranei.
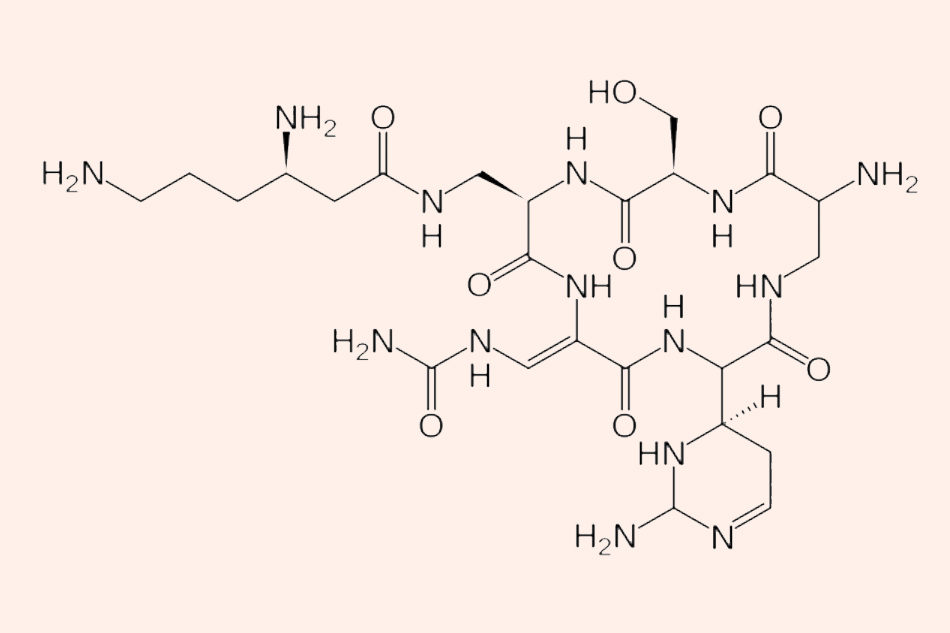
Kháng sinh này có nguồn gốc tự nhiên, được phân lập từ xạ khuẩn Streptomyces capreolus.
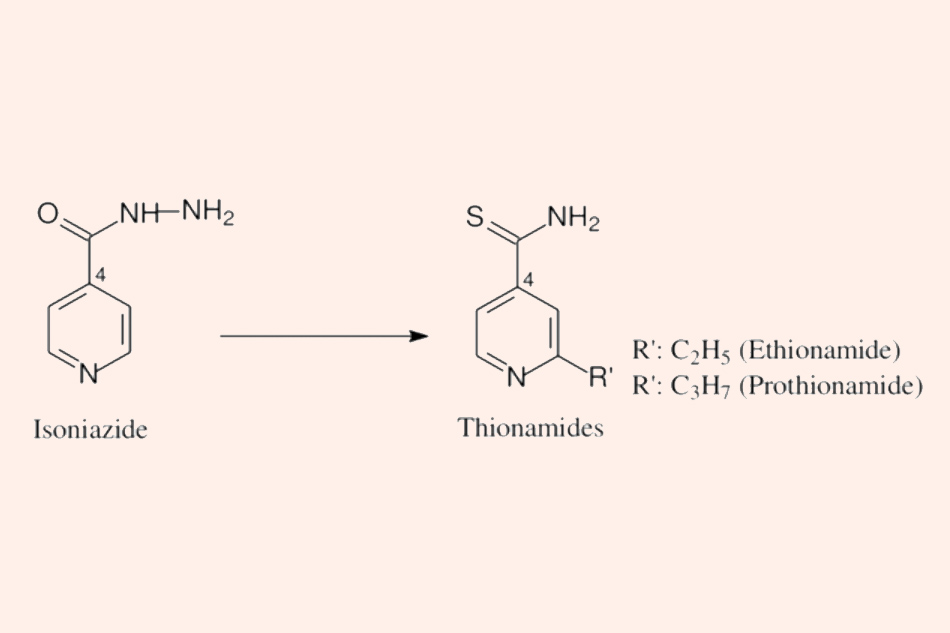
Cấu trúc hóa học của Ethionamide và Prothionamide (phải) và sự liên quan về cấu trúc hóa học với Isoniazid (trái).
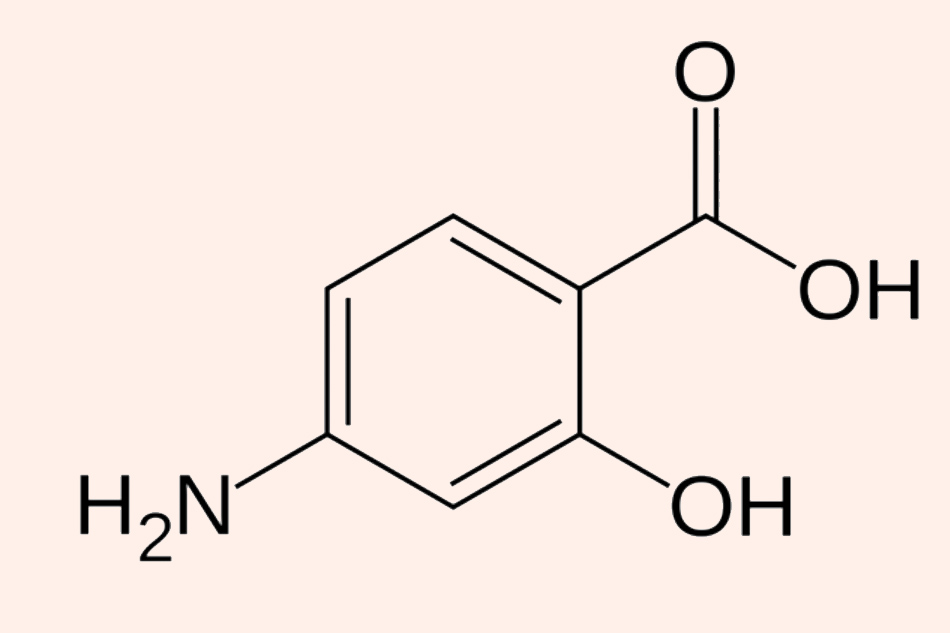
Đây là cấu trúc hóa học đơn giản của Para-aminosalicylic acid (PAS). Cấu trúc này có sự tương đồng với các Sulfamide kháng khuẩn.
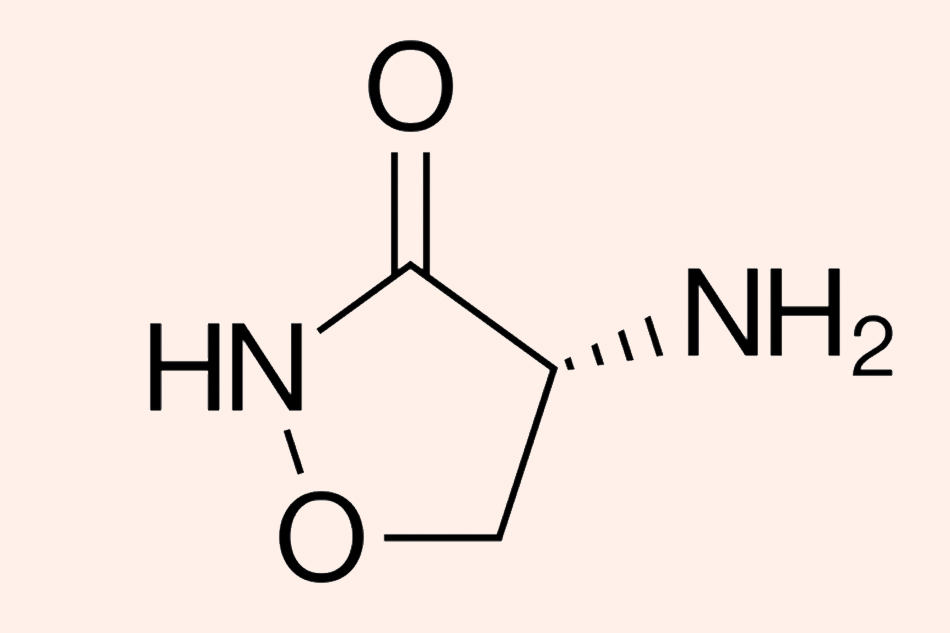
Đây là cấu trúc hóa học đơn giản của Cycloserine.
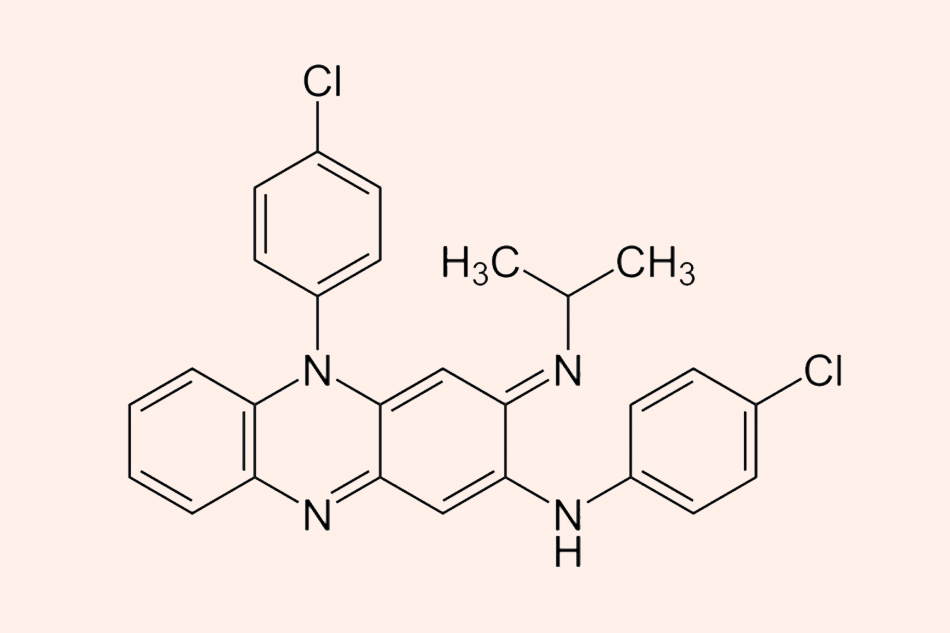
Đây là cấu trúc hóa học của Clofazimine.
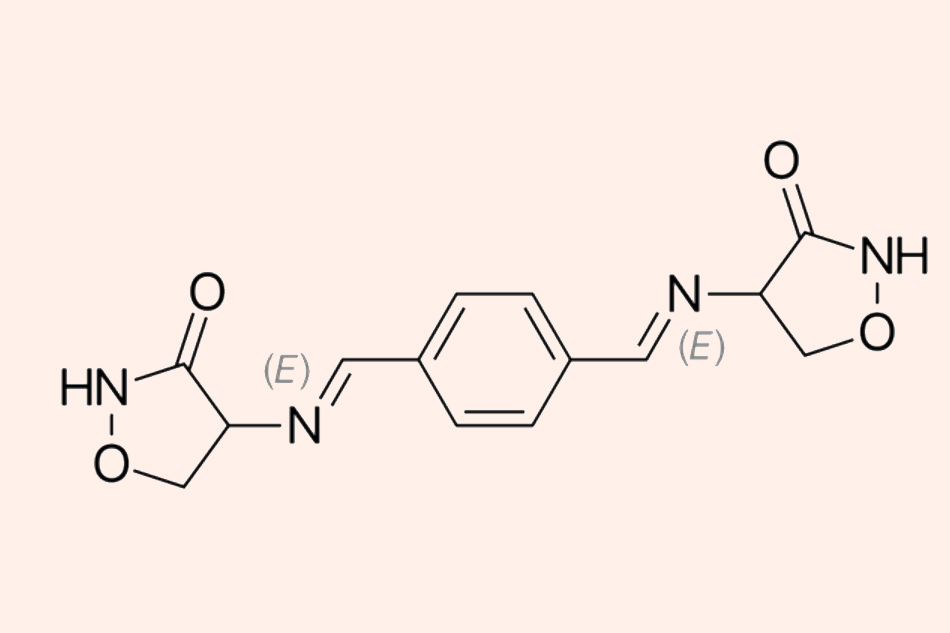
Đây là cấu trúc hóa học của Terizidone. Terizidone là một dẫn chất của Cycloserine.
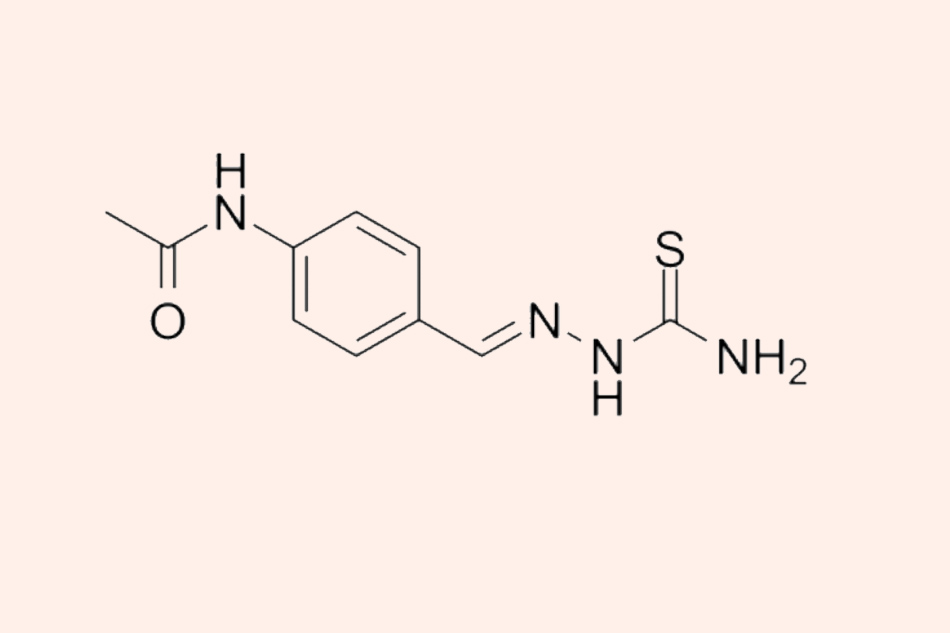
Đây là cấu trúc hóa học của Thiacetazone. Hiện nay thuốc này gần như không còn được sử dụng nữa do độc tính của nó, cũng như đã có các thuốc chống lao tốt hơn.
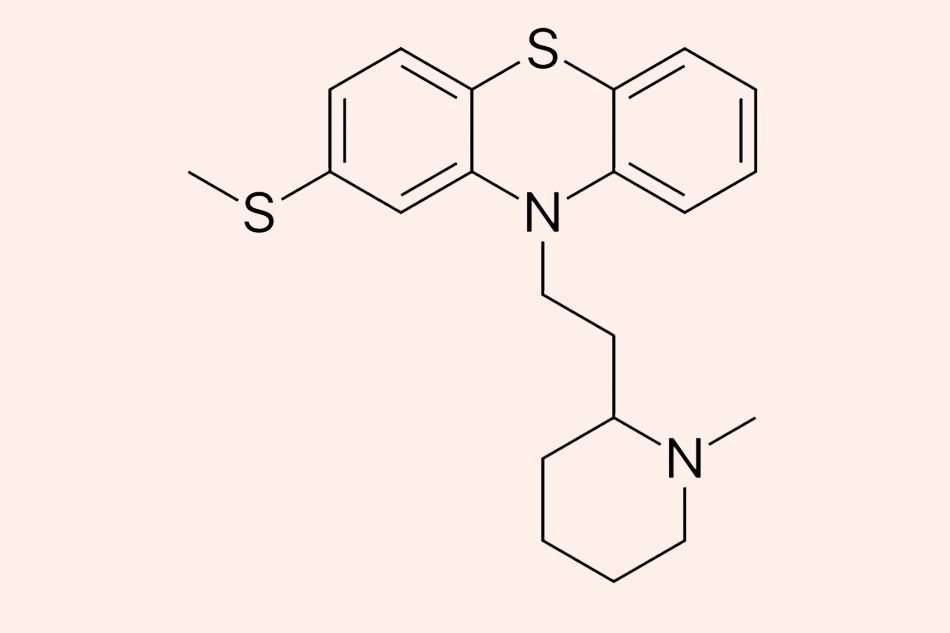
Đây là cấu trúc hóa học của Thioridazine. Ngoài tác dụng chống lao, Thioridazine còn được biết đến là một thuốc chống loạn thần thế hệ một nhóm Phenothiazine.
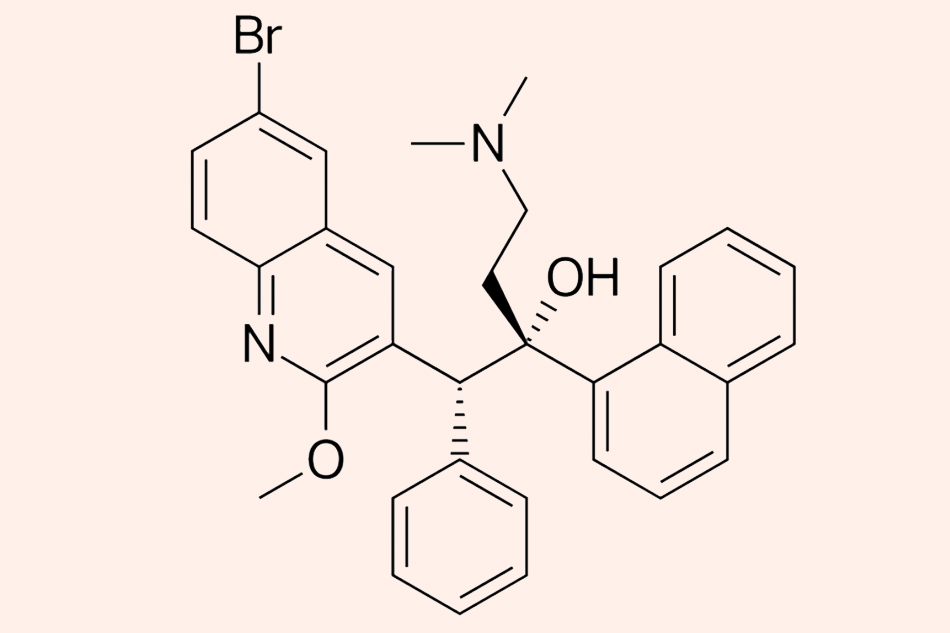
Đây là cấu trúc hóa học của Bedaquiline.
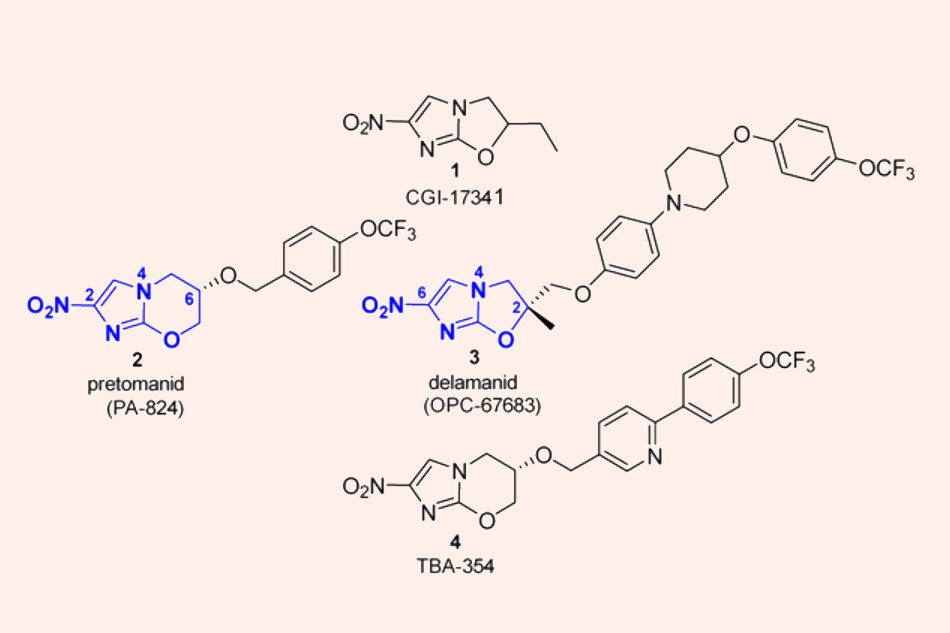
Đây là cấu trúc hóa học của Delamanid, Pretomanid và TBA-354, các thuốc thuộc nhóm Nitroimidazole.
Dược lý học
Dược lực học
Cơ chế tác dụng
- Isoniazid: Thuốc này được khuếch tán thụ động qua màng tế bào vi khuẩn lao. Isoniazid là một tiền thuốc (prodrug) và bản thân nó không độc với tế bào vi khuẩn lao. Tuy nhiên, nó có thể được chuyển thành dạng hoạt động bên trong tế bào vi khuẩn nhờ một enzyme của chính nó có tên là KatG. KatG là một enzyme catalase-peroxidase đa chức năng, nó bao gồm cả các hoạt tính khác như peroxynitritase và NADH oxidase.
KatG hoạt hóa Isoniazid bằng cách peroxy hóa nó, tạo ra chất độc nội bào là gốc có khả năng phản ứng cao. Có nhiều chất oxy hóa hỗ trợ sự oxy hóa Isoniazid của KatG, bao gồm superoxide, hydrogen peroxide và các alkyl hydroperoxide đơn giản. Hydrogen peroxide được tạo ra bên trong vi khuẩn và là sản phẩm phụ của quá trình trao đổi chất hiếu khí. Một số mô hình oxy hóa khác được đề xuất cũng có khả năng hoạt hóa được Isoniazid, bao gồm “horseradish peroxidase” và thậm chí là cả ion mangan vô cơ.
Nếu thiếu vắng NAD+ hoặc NADP+, không có chất hóa học ổn định nào là dẫn chất của Isoniazid được tạo ra trong quá trình hoạt hóa in vitro. Điều này chứng tỏ hoạt tính của Isoniazid bắt nguồn từ các chất trung gian phản ứng là dẫn chất của nó. Chất trung gian phản ứng này tạo liên kết cộng hóa trị bền vững với protein mang nhóm acyl (AcpM) và KasA (một protein mang nhóm β-ketoacyl synthetase). Khi các protein mang nhóm acyl này bị ức chế, sự tổng hợp acid mycolic (một thành phần quan trọng trong thành tế bào vi khuẩn lao) cũng bị ức chế.
Ngoài sự ức chế tổng hợp acid mycolic, các gốc tự do sinh ra từ Isoniazid cũng có khả năng làm hỏng các thành phần khác của tế bào như protein, lipid và nucleic acid. Ngoài các gốc tự do thông thường, gốc NO• cũng có thể đóng góp vào cơ chế của Isoniazid. Ở các chủng vi khuẩn lao có mycothiol, gốc NO• sẽ phản ứng với mycothiol tạo ra S‐nitrosomycothiol, chất này độc với tế bào vi khuẩn. Một số vi khuẩn sẽ sản xuất ra enzyme S‐nitrosomycothiol reductase để giải độc nó.
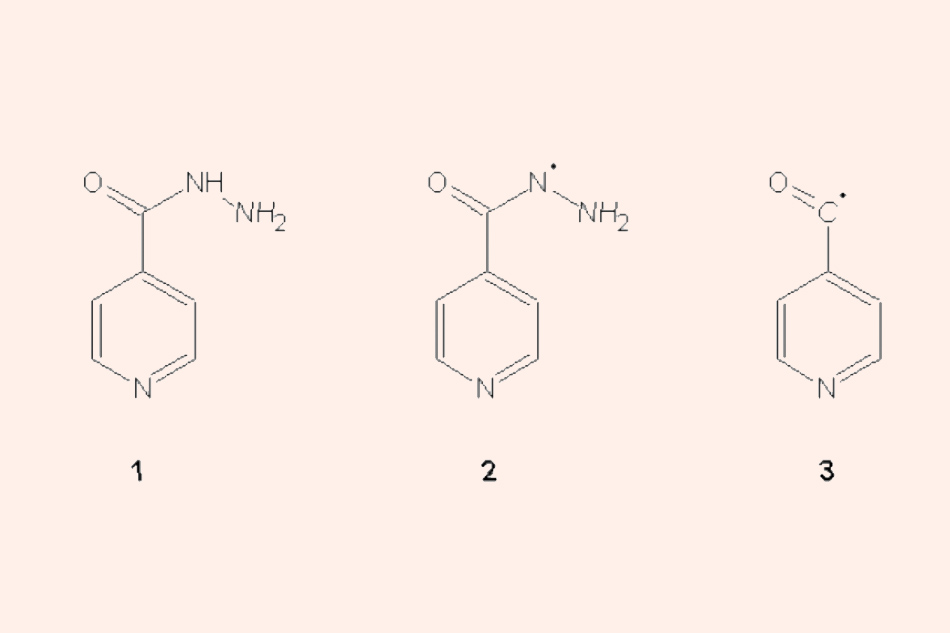
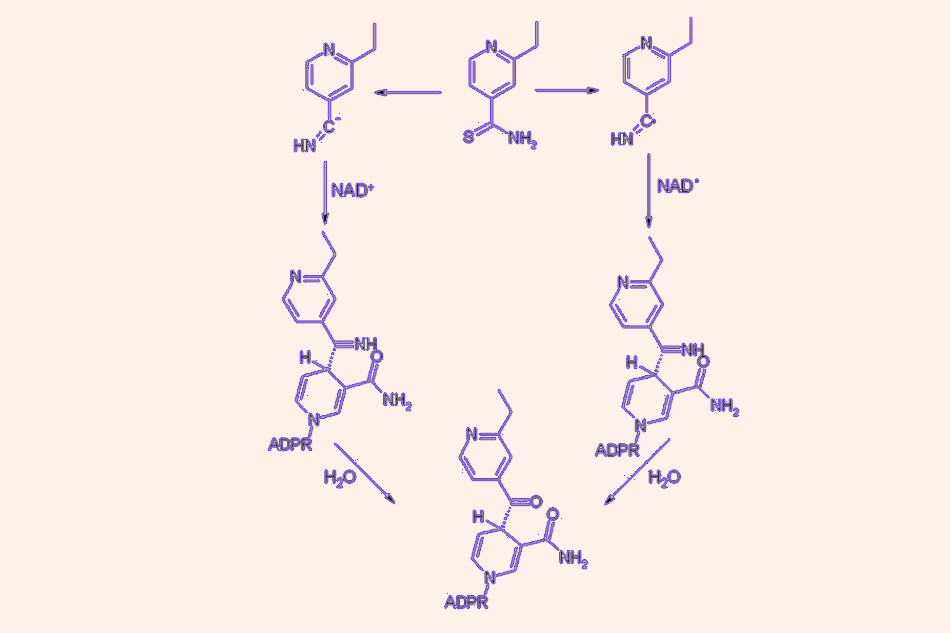
(1) Phân tử Isoniazid. (2) Gốc isonicotinic hydrazyl. (3) Gốc isonicotinoyl (hay còn gọi là gốc isonicotinic acyl).
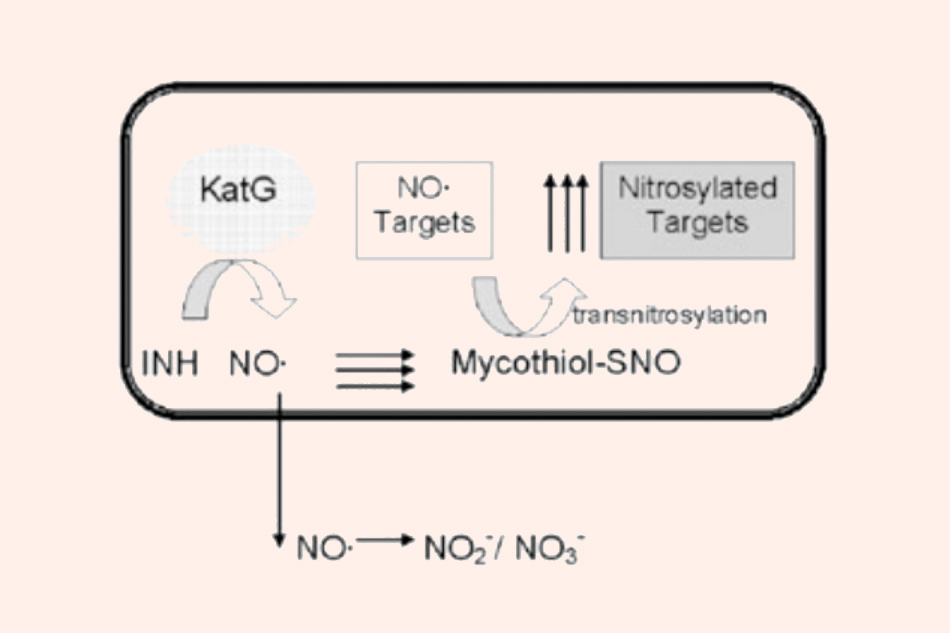
Phản ứng nội bào của gốc NO• sinh ra từ Isoniazid với mycothiol tạo ra S‐nitrosomycothiol. Nếu như không có mycothiol, hầu hết gốc NO• sinh ra sẽ được khuếch tán ra khỏi tế bào và bị oxy hóa trong môi trường xung quanh.
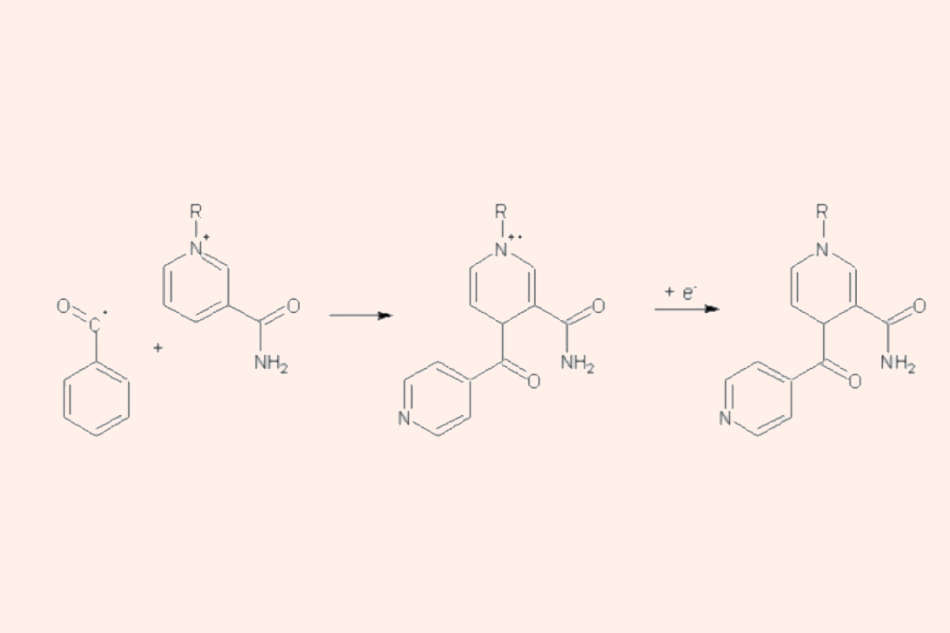
Sự hình thành của phức hợp Isoniazid-NAD(P)+ bằng phản ứng giữa gốc isonicotinoyl với NAD(P)+. Gốc R là adenosine diphosphoribose (NAD+) hoặc phosphoadenosine diphosphoribose (NADP+).
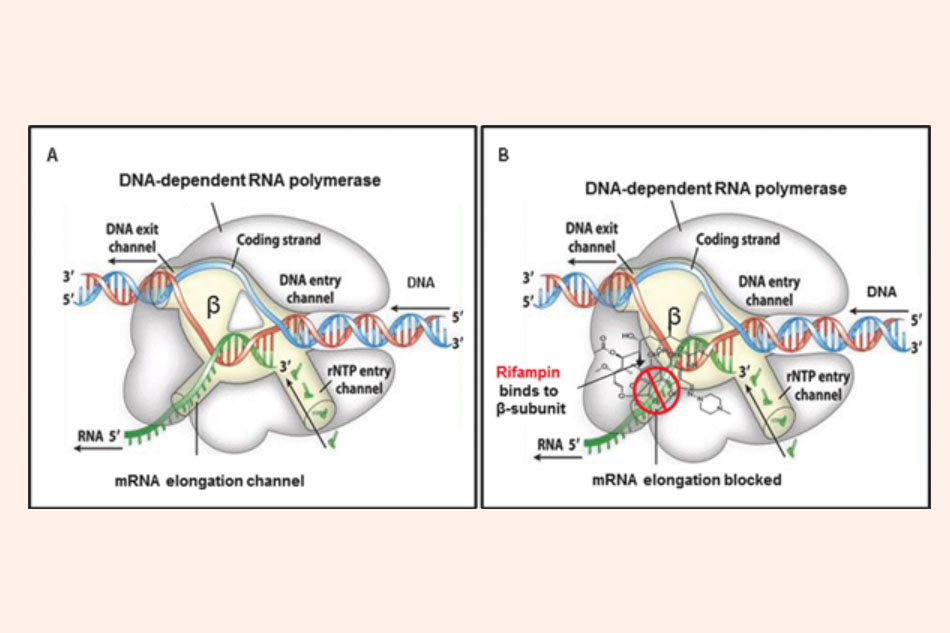
- Rifampicin: Rifampicin có khả năng gắn vào tiểu đơn vị β của RNA polymerase phụ thuộc DNA, từ đó ức chế RNA polymerase. Enzyme này có khả năng polymer hóa ribonucleoside triphosphates trên DNA và xúc tác quá trình phiên mã DNA thành RNA. Ức chế hoạt tính của enzyme này làm cho quá trình phiên mã của vi khuẩn bị ức chế. Rifampicin không ức chế RNA polymerase của người ở liều điều trị.
Ngoài vi khuẩn lao, thuốc cũng có tác dụng trên nhiều vi khuẩn gram dương khác, đặc biệt là tụ cầu vàng Staphylococcus aureus. Tác dụng của kháng sinh này trên vi khuẩn gram âm kém hơn nhiều, không phải do sự khác biệt trong độ nhạy cảm của RNA polymerase với phân tử kháng sinh, mà là bởi khả năng thấm qua lớp màng ngoài vi khuẩn gram âm của Rifampicin khá hạn chế.
- Pyrazinamide: Bản thân Pyrazinamide không có tác dụng trên vi khuẩn lao. Tác dụng điều trị lao của Pyrazinamide là thông qua chất chuyển hóa Pyrazinoic acid, quá trình chuyển hóa này được thực hiện nhờ enzyme pyrazinamidase của trực khuẩn lao. Enzyme này được mã hóa bởi gene pncA. Pyrazinoic acid làm cản trở chức năng vận chuyển và chuyển hóa của màng tế bào vi khuẩn.
Pyrazinamide bị bất hoạt ở pH trung tính và có tác dụng ở pH hơi acid, cụ thể là pH 5.5 trong lysosome của đại thực bào. Do vậy, Pyrazinamide không có tác dụng với các vi khuẩn lao ở ngoại bào mà chỉ có tác dụng trên các vi khuẩn lao cư trú trong lysosome của đại thực bào.
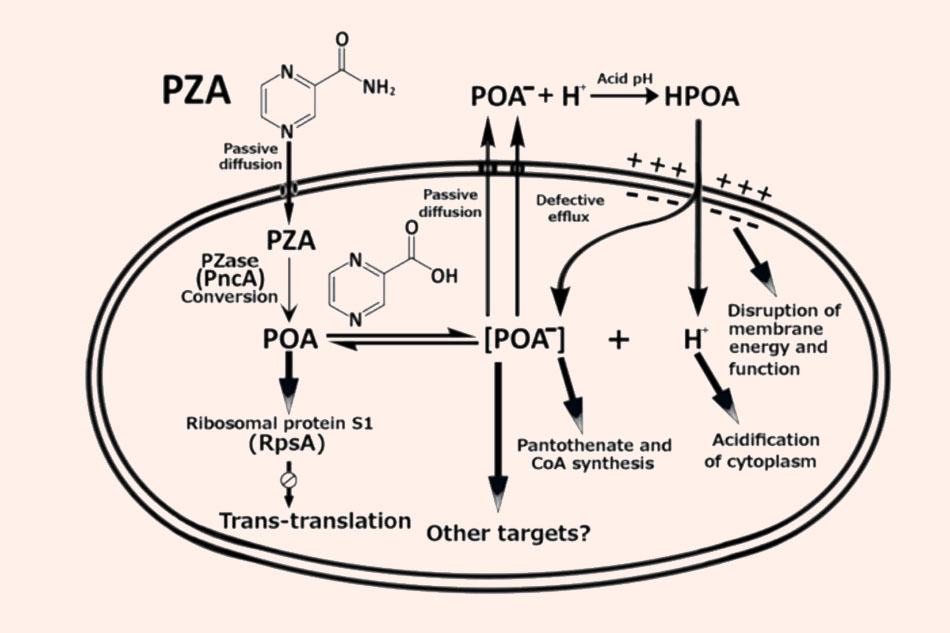
Con đường đi và chuyển hóa của Pyrazinamide trong trực khuẩn lao cùng với cơ chế tác dụng của nó. Pyrazinamide được chuyển hóa thành Pyrazinoic acid nhờ enzyme của vi khuẩn. Pyrazinoic acid dạng anion hóa sau đó được vận chuyển ra khỏi tế bào nhờ khuếch tán thụ động hoặc bơm đẩy. Pyrazinoic acid sau đó đi từ ngoại bào vào tế bào chất sẽ làm acid hóa tế bào chất, ức chế hoạt tính của một số enzyme, ngoài ra nó cũng làm giảm chênh lệch nồng độ proton giữa hai bên màng tế bào và ảnh hưởng đến sự vận chuyển màng, ngăn cản tổng hợp protein và RNA. Nếu pH ngoại bào trung tính hoặc hơi kiềm, Pyrazinoic acid sẽ tồn tại ở dạng anion hóa, không thể qua được màng tế bào vi khuẩn và do đó không thể hiện tác dụng. Đây là lý do tại sao thuốc này chỉ thể hiện tác dụng trong lysosome của đại thực bào, khi pH môi trường hơi acid (pH 5.5).
- Ethambutol: Cơ chế tác dụng của Ethambutol chưa hoàn toàn rõ ràng. Có bằng chứng cho thấy thuốc ức chế enzyme arabinosyl transferase, enzyme chịu trách nhiệm polymer hóa arabinose thành arabinan và sau đó là arabinogalactan. Chất này là thành phần quan trọng của thành tế bào vi khuẩn lao. Thuốc có tác dụng kìm khuẩn.
Ethambutol cũng được cho là có tác dụng hiệp đồng với Isoniazid thông qua ức chế gene inhA.
- Capreomycin: Capreomycin là kháng sinh ức chế tổng hợp protein của vi khuẩn lao thông qua ức chế ribosome 70S của vi khuẩn. Tuy nhiên cơ chế thực sự ẩn sau điều này còn chưa rõ. Có bằng chứng cho thấy thuốc ức chế sự tương tác giữa protein L12 (được mã hóa bởi gene rplL) và L10 (được mã hóa bởi gene rplJ) trong tiểu đơn vị ribosome 50S. Một số nghiên cứu khác cho thấy Capreomycin có thể gắn vào cả tiểu đơn vị ribosome 30S và 50S, hoặc cản trở tRNA đã được deacyl hóa di chuyển từ vị trí A đến vị trí P, bỏ trống vị trí A cho tRNA aminoacyl tiếp theo.
- Ethionamide và Prothionamide: Các thuốc này có cấu trúc hóa học tương tự nhau và tương tự như Isoniazid, do đó cơ chế tác dụng cũng tương tự nhau, đó là ức chế sinh tổng hợp acid mycolic thành tế bào. Đích tác dụng chính của Ethionamide và Isoniazid đều là InhA, enoyl-acyl ACP reductase tham gia vào tổng hợp acid mycolic.
- Cycloserine: Cycloserine có cấu trúc tương tự D-alanine và ức chế sự tích hợp của D-alanine vào pentapeptide của peptidoglycan thành tế bào bằng cách ức chế cạnh tranh enzyme alanine racemase (enzyme này chịu trách nhiệm chuyển L-alanine thành D-alanine) và D-alanyl-D-alanine ligase.
- Para-aminosalicylic acid: Thuốc này ức chế tổng hợp folate của vi khuẩn. Cấu trúc của nó tương tự như Para-aminobenzoic acid (PABA), do vậy cơ chế tác dụng của nó được coi là tương tự như các Sulfamide kháng khuẩn.
- Clofazimine: Cơ chế hoạt động chính xác của Clofazimine chưa được hiểu rõ. Các nhà khoa học đã chứng minh được ở M. smegmatis, Clofazimine là tiền thuốc và được khử bởi NADH-quinone oxidoreductase type 2 (NDH-2) và giải phóng các loại oxy phản ứng khi tái oxy hóa tự nhiên từ O2. Người ta cho rằng Clofazimine cạnh tranh với menaquinone (MK-4; vitamin K2), quinone duy nhất hiện diện trong Mycobacteria và là một chất nhận điện tử quan trọng, được khử bởi NDH-2. MK-4 còn được biết đến với cái tên menatetrenone (C31H40O2), gồm một vòng quinone liên kết với một chuỗi 4 nhóm isoprenoid.
- Terizidone: Cơ chế tác dụng tương tự như Cycloserine. Terizidone là một dẫn xuất của Cycloserine.
- Thiacetazone: Các nghiên cứu cho thấy Thiacetazone ức chế các methyltransferase chịu trách nhiệm đưa vòng cyclopropane vào mycolate ở các loài M. bovis, M. chelonae và M. marinum. Các thành viên nằm trong họ enzyme methyltransferase này bao gồm PcaA, CmaA2 và MmaA2. Tuy rằng các enzyme này cũng khá quan trọng đối với vi khuẩn lao, nhưng việc ức chế chúng chưa thể giải thích được cho cơ chế chống lao của Thiacetazone. Thiacetazone ức chế sinh tổng hợp acid mycolic ở M. tuberculosis và M. kansasii.
- Thioridazine: Thioridazine là chất đối kháng calmodulin và hoạt tính chống trực khuẩn lao của thuốc có liên quan đến sự hiện diện của một protein giống calmodulin trong vi khuẩn. Không chỉ có tác dụng trên các chủng trực khuẩn lao kháng thuốc, thuốc này còn có tác dụng trên cả các trực khuẩn cư trú trong đại thực bào. Cơ chế đầy đủ của thuốc còn chưa hoàn toàn được hiểu rõ. Liên quan đến sự ức chế vận chuyển calcium và các enzyme phụ thuộc calcium, nó cũng ức chế sự tạo ra năng lượng của tế bào do thủy phân ATP. Thioridazine cũng được cho là ức chế các bơm tống thuốc giả định. Gần đây, Thioridazine còn được chứng minh làm ảnh hưởng đến mạng lưới yếu tố sigma của M. tuberculosis, mạng lưới này đóng vai trò trong việc bảo vệ vi khuẩn chống lại các tổn thương tế bào.
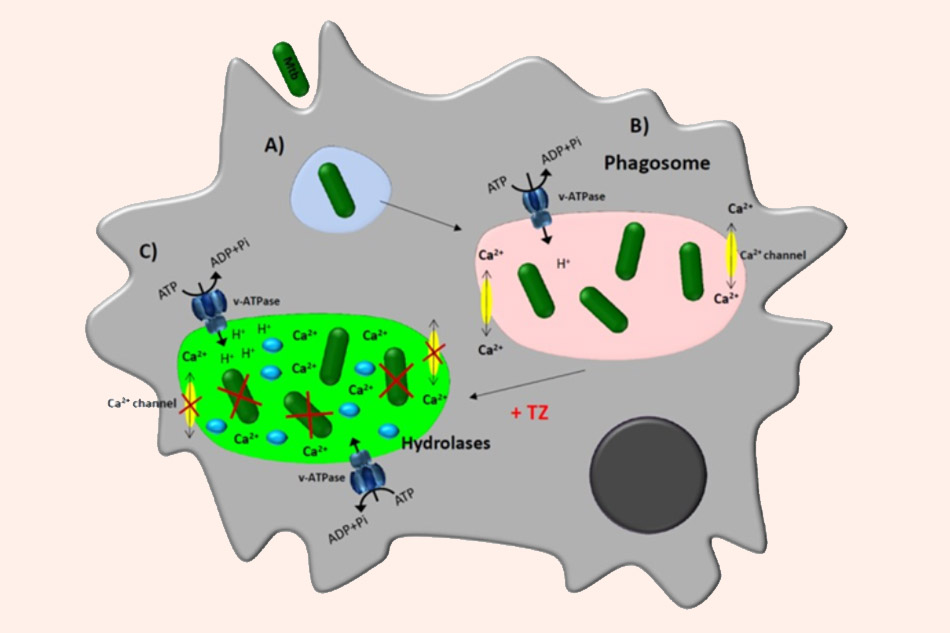
Cơ chế được đề xuất cho hoạt động chống lại trực khuẩn lao cư trú trong đại thực bào của Thioridazine. (A) Trực khuẩn xâm nhập vào đại thực bào và nằm trong phagosome. (B) Trong phagosome, trực khuẩn điều khiển đáp ứng miễn dịch, làm giảm lượng calcium có sẵn trong phagosome, cản trở quá trình acid hóa cần thiết để hoạt hóa hydrolase, do đó vi khuẩn không bị giết. (C) Điều trị bằng các thuốc chẹn kênh ion Ca2+ như Thioridazine làm tăng nồng độ calcium trong tế bào chất, cùng với đó là sự phiên mã và hoạt động của H+-ATPase không bào. Điều này dẫn đến làm tăng nồng độ proton trong phagosome, hoạt hóa hydrolase và tiêu diệt trực khuẩn lao.
- Bedaquiline: Thuốc làm bất hoạt chọn lọc F1/F0-ATP synthase của trực khuẩn lao, nhưng không ức chế F1/F0-ATP synthase ở động vật có vú. Đây là một enzyme quan trọng của vi khuẩn vì nó giúp sản xuất ra ATP. Thành phần F0 thân dầu trong F1/F0-ATP synthase, đặc biệt là tiểu đơn vị oligomer xuyên màng C (AtpE), là đích tác dụng chính của Bedaquiline. Liên kết của Bedaquiline nằm trong “khe hở giữa hai tiểu đơn vị C liền kề trong vòng C”, vùng chứa Glu-61 gắn proton. Hệ quả là thuốc làm cản trở sự di chuyển của proton và sự tổng hợp ATP.
Mặc dù sau khi phơi nhiễm Bedaquiline, có sự giảm nhanh ATP trong vi khuẩn lao, nhưng vi khuẩn không bị tiêu diệt ngay mà phải mất vài ngày, nguyên nhân có thể là do cảm ứng trạng thái ngủ của vi khuẩn hoặc nó sử dụng các con đường sinh năng lượng thay thế. Ngoài cơ chế chính là ức chế tổng hợp ATP, thuốc còn có những cơ chế khác vẫn đang được nghiên cứu hoặc đang gây tranh cãi.
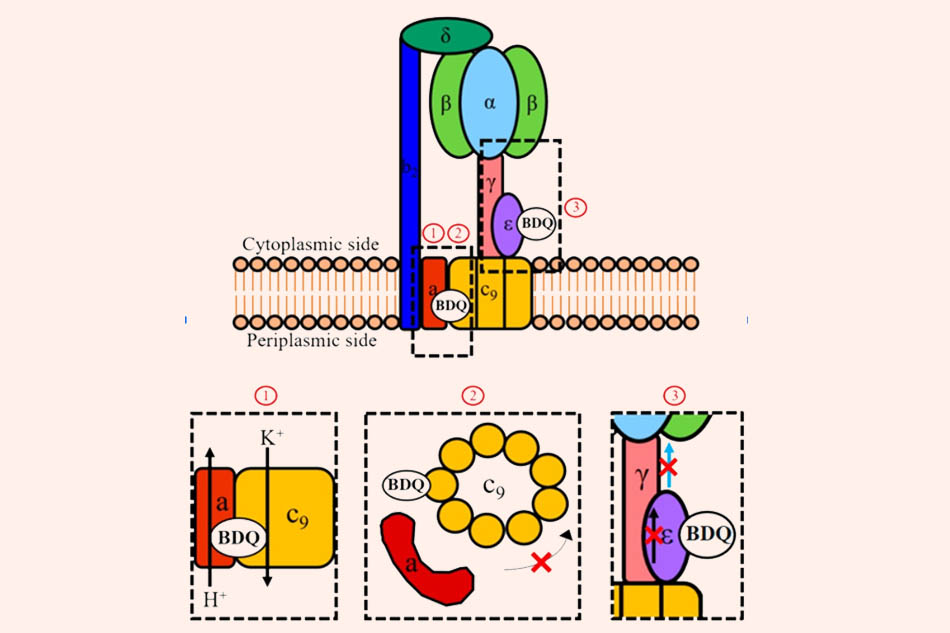
Đây là cơ chế hoạt động của Bedaquiline (BDQ). Hình ảnh biểu diễn phức hợp enzyme F-ATP synthase ở trực khuẩn lao. Trong quá trình tổng hợp ATP, sự chênh lệch nồng độ proton thúc đẩy sự quay của vòng c (c9). Dòng proton đi qua mặt phân cách giữa tiểu đơn vị a và vòng c. Chuyển động quay này được truyền bởi các tiểu đơn vị γ- và ε- đến phần đầu α3β3, nơi thúc đẩy tổng hợp ATP. Cơ chế hoạt động gián tiếp (1) của thuốc là chống vận chuyển H+/K+ từ khu vực xung quanh của vị trí gắn vòng c của nó, do đó làm giảm chênh lệch nồng độ proton, ngăn không cho vòng c quay. Điều này tách rời sự vận chuyển điện tử trong chuỗi truyền điện tử từ quá trình tổng hợp ATP tại F-ATP synthase. Các cơ chế hoạt động trực tiếp của thuốc là nhắm mục tiêu vòng c (2) và tiểu đơn vị ε của enzyme (3). Liên kết với tiểu đơn vị c làm cản trở sự quay của vòng c, do đó ức chế tổng hợp ATP tại phần đầu α3β3. Liên kết với tiểu đơn vị ε làm cản trở mạng lưới tương tác amino acid trong tiểu đơn vị, từ đó cản trở sự truyền tin về chuyển động quay của vòng c trong toàn tiểu đơn vị và đến đầu α3β3.
- Delamanid và Pretomanid: Các thuốc này là dẫn xuất hóa học của dihydro-nitroimidazooxazole. Chúng ức chế tổng hợp thành phần quan trọng của thành tế bào vi khuẩn lao, đó là methoxy mycolic acid và ketomycolic acid (thuốc không ức chế α-mycolic acid như Isoniazid). Chúng đều là một tiền thuốc (prodrug), chỉ hoạt động khi được hoạt hóa bằng enzyme nitroreductase phụ thuộc deazaflavin (Rv3547). Chất chuyển hóa trung gian được tạo thành giữa Delamanid và dẫn xuất desnitro-imidazooxazole đóng vai trò quan trọng trong ức chế tổng hợp mycolic acid.
Cơ chế đề kháng
- Isoniazid: Cơ chế đề kháng với Isoniazid có thể là đột biến dẫn đến biểu hiện quá mức gene inhA, mã hóa một protein mang nhóm acyl reductase phụ thuộc NADH, hoặc đột biến (có thể xóa bỏ) gene quy định KatG, hoặc đột biến vùng khởi động dẫn đến biểu hiện quá mức ahpC, một gene giúp bảo vệ tế bào vi khuẩn lao khỏi stress oxy hóa, hay đột biến ngay chính gene mã hóa cho KasA. Trong đó, tăng biểu hiện gene inhA không gây ra đề kháng với Isoniazid cao (và do đó có thể xử trí bằng cách tăng liều thuốc) nhưng gây ra đề kháng chéo với Ethionamide. Đột biến KatG sẽ dẫn đến kháng Isoniazid ở mức độ cao (không thể giải quyết bằng tăng liều thuốc) nhưng lại không dẫn đến đề kháng chéo với Ethionamide.
Cơ chế đề kháng Isoniazid phổ biến nhất là đột biến thay đổi KatG. Những đột biến này thường là các đột biến điểm chọn lọc tạo ra protein có hoạt tính một phần, các hoạt tính này vẫn giúp duy trì sự tồn tại và phát triển của trực khuẩn lao nhưng làm giảm độc tính của Isoniazid với vi khuẩn.
Tần suất xuất hiện đột biến kháng Isoniazid trong quần thể trực khuẩn lao nhạy cảm là cứ một triệu vi khuẩn thì xuất hiện một vi khuẩn đột biến, tức 10-6.
- Rifampicin: Cơ chế đề kháng với Rifampicin phổ biến nhất là đột biến điểm gene rpoB, gene mã hóa cho tiểu đơn vị β của RNA polymerase. Đột biến này làm giảm ái lực gắn của kháng sinh với đích tác dụng của nó. Tần suất xuất hiện đột biến kháng Rifampicin cũng tương đương Isoniazid.
Cơ chế khác đề kháng với Rifampicin bao gồm giảm khả năng kháng sinh tiếp cận được đích tác dụng bằng cách giảm tính thấm màng tế bào. Cơ chế này phổ biến ở vi khuẩn gram âm.
- Pyrazinamide: Cơ chế đề kháng với Pyrazinamide có thể là giảm tính thấm màng tế bào của thuốc hoặc đột biến gene pncA, từ đó làm giảm khả năng chuyển Pyrazinamide thành dạng hoạt động Pyrazinoic acid. Phần lớn các đột biến ở pncA là các đột biến sai lệch gây ra thay thế amino acid, và trong một số trường hợp là chèn hoặc xóa các nucleotide, đột biến vô nghĩa trong cấu trúc gene pncA hoặc đột biến vùng promoter giả định của pncA. Đột biến các vị trí C138, D8, K96, D49, H51 và H71 làm thay đổi bộ ba vị trí hoạt động và vị trí liên kết với kim loại. Các phần F13, L19, H57 (vị trí đột biến đặc trưng H57D trong M. bovis), W68, G97, Y103, I113, A134 và H137 đột biến cũng được dự đoán là gây mất hoạt tính enzyme. Đột biến ở Q10, D12, S104 và T142 được dự đoán cũng gây cản trở tương tác liên kết hydro giữa các nguyên tử chuỗi bên và chuỗi chính.
Ngoài đột biến gene pncA, một số đột biến gene khác như đột biến rpsA và panD cũng có liên quan đến kháng Pyrazinamide. Đột biến rpsA xuất hiện ở M. canettii gây ra đề kháng thuốc mức độ thấp. Các đột biến rpsA có thể gặp bao gồm Thr5Ala, Pro9Pro, Thr210Ala và Glu457Glu. Đột biến gene panD (mã hóa cho enzyme aspartate decarboxylase) cũng liên quan đến kháng Pyrazinamide nhưng không phổ biến. Các đột biến panD đã được xác định trong các chủng M. canettii kháng Pyrazinamide được phân lập từ tự nhiên và một chủng M. tuberculosis đa kháng thuốc được phân lập trên lâm sàng. panD tham gia vào quá trình tổng hợp β-alanine, đây là tiền chất cho sinh tổng hợp pantothenate và co-enzyme A. Các chất này rất quan trọng với sự tồn tại của vi khuẩn.
Tần suất xảy ra đột biến đề kháng Pyrazinamide cao hơn so với Isoniazid và Rifampicin, bằng khoảng 10-5.
- Ethambutol: Operon embCAB mã hóa cho arabinosyl transferase. Đề kháng Ethambutol thường là do đột biến biểu hiện quá mức sản phẩm của gene emb hoặc đột biến cấu trúc gene embB. Khi giá trị MIC (nồng độ ức chế tối thiểu) của vi khuẩn lao vượt quá 5 µg/mL thì được coi là kháng Ethambutol.
Tần suất xuất hiện đột biến kháng Ethambutol là 10-7.
- Capreomycin: Không có nhiều nghiên cứu về cơ chế đề kháng của trực khuẩn lao với Capreomycin. Đột biến gene tlyA (Rv1694) trên genome H37Rv của M. tuberculosis có liên quan đến kháng Capreomycin. Một số đột biến gene khác cũng liên quan đến đề kháng Capreomycin là các gene rrs và eis.
- Ethionamide và Prothionamide: Đột biến một acid amin S94A của inhA gây ra đề kháng cả Ethionamide và Isoniazid. Hoặc biểu hiện quá mức gene inhA cũng gây ra kháng cả hai loại thuốc này. Đột biến gene ethA (mã hóa cho một flavin monooxygenase xúc tác cho phản ứng Baeyer-Villiger để giải độc các ketone thơm chuỗi dài) cũng cho thấy khả năng đề kháng với Ethionamide.
Nếu vi khuẩn lao đề kháng Ethionamide, nó cũng sẽ đề kháng với Prothionamide.
- Cycloserine: Biểu hiện quá mức Alr gây ra đề kháng với Cycloserine. Alr là đích tác dụng chính của thuốc ở M. smegmatis. Biểu hiện quá mức gene mã hóa cho alanine racemase ở M. smegmatis cũng gây ra kháng Cycloserine. Đột biến chính xác gây ra kháng thuốc ở M. tuberculosis chưa được xác định.
- Para-aminosalicylic acid: Đề kháng với thuốc chủ yếu liên quan đến enzyme thymidylate synthase A (ThyA). Đây là enzyme đóng vai trò quan trọng trong sinh tổng hợp folate của trực khuẩn lao. Đột biến ở các phần cấu trúc quan trọng của ThyA liên quan đến tương tác với cơ chất 2′-deoxyuridine-5′-monophosphate (dUMP), cofactor 5,10-methylenetetrahydrofolate (MTHF) và vị trí xúc tác gây ra kháng thuốc ở bệnh nhân lao.
- Clofazimine: Locus rv0678 có liên quan đến kháng thuốc in vitro cũng như kháng thuốc trên lâm sàng (mã hóa cho bơm tống thuốc). Protein Rv0678 có vai trò như một bộ điều hòa phiên mã, ức chế biểu hiện của mmpS5-mmpL5, gene mã hóa cho bơm tống đa thuốc MmpS5-MmpL5. Đột biến này còn có thể dẫn đến kháng chéo với Bedaquiline. Tuy vậy, locus này không có ở M. leprae (gây bệnh phong), và hiện tại vi khuẩn này vẫn rất nhạy cảm với Clofazimine. Trong vài năm trở lại đây, đột biến ở locus rv1979c và rv2535c cũng cho thấy sự liên quan đến đề kháng các riminophenazine.
- Terizidone: Cơ chế đề kháng của thuốc chưa được hiểu rõ, nhưng có lẽ sẽ tương tự như Cycloserine do chúng có cùng cơ chế tác dụng. Vi khuẩn sản xuất quá mức alanine cũng có thể làm giảm khả năng ức chế tổng hợp thành tế bào của thuốc.
- Thiacetazone: Các chủng M. bovis kháng Thiacetazone có đột biến thiếu keto-mycolate đã được xác định. Chúng sở hữu đột biến trong gene mmaA4, gene này mã hóa cho enzyme methyltransferase cần thiết cho sự tổng hợp các methoxy- và keto-mycolic acid. Ngoài ra, đột biến ở gene ethA hoặc một số gene khác cũng cho thấy đề kháng với Thiacetazone. Đột biến operon hadABC cũng cho thấy liên quan đến đề kháng Thiacetazone ở M. tuberculosis và M. kansasii. Phức hợp FAS-II dehydratase có thể là một nhân tố mới tạo ra sự đề kháng ở M. tuberculosis do điều trị bằng Thiacetazone dẫn đến sự tích tụ các acid béo 3-hydroxy, cơ chất của dehydratase.
- Thioridazine: Có rất ít nghiên cứu về đề kháng Thioridazine ở vi khuẩn lao. Nhưng trên lý thuyết, việc xuất hiện đột biến kháng thuốc là khá khó do đột biến gene mã hóa protein vận chuyển calcium tương tự calmodulin có thể dẫn đến tổn thương nghiêm trọng cho chính bản thân vi khuẩn. Ngoài ra, thuốc cũng ảnh hưởng đến nhiều mục tiêu cần thiết cho sự phát triển của vi khuẩn, do đó ngay cả khi đột biến xảy ra, xác suất để tất cả các đột biến có thể xảy ra trong một tế bào đơn lẻ là rất khó.
- Bedaquiline: Một số cơ chế đề kháng với Bedaquiline đã được xác định, trong đó nổi bật nhất là đột biến ở hai gene riêng biệt. Đầu tiên là đột biến ở gene atpE, mã hóa cho F1/F0-ATP synthase. Khoảng 30% các chủng lâm sàng kháng Bedaquiline có đột biến này. Loại đề kháng này chỉ liên quan đến đột biến ở tiểu đơn vị C của enzyme. Gene thứ hai có liên quan đến kháng thuốc là rv0678, mã hóa cho protein Rv0678. Đột biến này được phát hiện rất phổ biến ở các vi khuẩn kháng Bedaquiline. Đột biến gene cũng dẫn đến kháng chéo với Clofazimine như đã nói ở trên.
Một cơ chế đề kháng với Bedaquiline khác cũng có thể góp phần gây ra thất bại điều trị, đó là hoạt hóa các bơm tống thuốc. Các chất ức chế bơm tống thuốc thông qua làm giảm điện thế xuyên màng có thể làm tăng tính nhạy cảm của vi khuẩn với thuốc, ví dụ như Verapamil, Reserpine và Valinomycin.
Tần suất phát triển đề kháng với Bedaquiline là 10-8.
- Delamanid: Đột biến một trong năm gene F420 coenzyme, bao gồm fgd, Rv3547, fbiA, fbiB và fbiC có liên quan đến đề kháng Delamanid và Pretomanid. Các nhà khoa học cho rằng không có sự đề kháng chéo giữa Delamanid và các thuốc chống lao khác, trừ Pretomanid.
Dược động học
Isoniazid
- Hấp thu: Hấp thu nhanh và hoàn toàn qua đường uống.
- Phân bố: Phân bố tốt vào các mô và dịch trong cơ thể, kể cả dịch não tủy. Có khả năng đi qua nhau thai và sữa mẹ. Liên kết với protein huyết tương thấp.
- Chuyển hóa: Thuốc trải qua các phản ứng acetyl hóa, thủy phân và liên hợp glycine tại gan. Đặc biệt phản ứng acetyl hóa có tính chất đa hình di truyền, với hai kiểu hình là acetyl hóa nhanh và acetyl hóa chậm. Các chất chuyển hóa được tạo thành bao gồm acetyl isoniazid, isonicotinic acid, isonicotinuric acid, isonicotinoyl-hydrazone của acid pyruvic và acid glutaric, N-methylisoniazid.
- Thải trừ: Thời gian bán thải (t1/2) ở những người có kiểu hình acetyl hóa nhanh là 1.2 giờ và kiểu hình acetyl hóa chậm là 3.5 giờ. Hơn 90% liều được bài xuất qua thận trong 24 giờ. Bài xuất qua phân không quá 10%.
Rifampicin
- Hấp thu: Hấp thu nhanh qua đường uống khi dạ dày rỗng. Dùng cùng thức ăn làm giảm hấp thu thuốc.
- Phân bố: Phân bố rộng vào các mô và dịch trong cơ thể, kể cả dịch não tủy. Liên kết với protein huyết tương 80%.
- Chuyển hóa: Thuốc được chuyển hóa tại gan. Chất chuyển hóa còn hoạt tính.
- Thải trừ: t1/2 là 3 giờ sau một liều 600 mg và 5.1 giờ sau một liều 900 mg. Nếu dùng lặp lại, thời gian bán thải giảm và đạt ngưỡng cân bằng 2-3 giờ. Thuốc được bài xuất chủ yếu vào mật và có chu kỳ gan – ruột. 30% liều được bài xuất qua nước tiểu.
Pyrazinamide
- Hấp thu: Hấp thu nhanh qua đường uống. Thời gian đạt nồng độ đỉnh trong huyết tương (Tmax) khoảng 2 giờ.
- Phân bố: Phân bố tốt vào các mô và dịch trong cơ thể. Thuốc qua được hàng rào máu não khi màng não bị viêm.
- Chuyển hóa: Chuyển hóa tại gan.
- Thải trừ: t1/2 khoảng 9-10 giờ. Khoảng 30% liều được bài xuất qua nước tiểu dưới dạng chuyển hóa acid pyrazinoic và 4% dưới dạng không đổi.
Ethambutol
- Hấp thu: Hấp thu nhanh qua đường uống và giảm không đáng kể khi dùng cùng thức ăn. Tmax khoảng 4 giờ.
- Phân bố: Phân bố tốt vào hồng cầu và qua được hàng rào máu não khi màng não bị viêm. Thuốc cũng qua được nhau thai.
- Chuyển hóa: Chủ yếu được oxy hóa bởi aldehyde dehydrogenase tại gan, sau đó chuyển thành dẫn chất acid dicarboxylic là 2,2′-(ethylinediimino)di-butyric acid.
- Thải trừ: 50% liều được bài xuất dưới dạng không đổi qua nước tiểu (8-15% dưới dạng chất chuyển hóa không hoạt động) và 20% liều được bài xuất qua phân trong vòng 48 giờ. t1/2 là 3.3 giờ ở bệnh nhân có chức năng thận bình thường và có thể kéo dài đến 7 giờ hoặc lâu hơn ở bệnh nhân suy thận.
Capreomycin
- Hấp thu: Tiêm bắp hấp thu gần như hoàn toàn. Tmax khoảng từ 1-2 giờ.
- Phân bố: Không có dữ liệu.
- Chuyển hóa: Không có dữ liệu.
- Thải trừ: Với liều 1 g, 52% liều được bài xuất qua nước tiểu trong vòng 12 giờ.
Ethionamide và Prothionamide
- Hấp thu: Hấp thu hoàn toàn sau khi uống, hầu như không có chuyển hóa bước đầu qua gan. Tmax của Ethionamide khoảng 1 giờ và Prothionamide khoảng 2 giờ.
- Phân bố: Phân bố tốt vào các mô và dịch trong cơ thể. Cả hai thuốc đều vào được dịch não tủy đáng kể. Liên kết với protein huyết tương của Ethionamide khoảng 30%.
- Chuyển hóa: Ethionamide được chuyển hóa rộng ở gan thành các chất có hoặc không có hoạt tính. Sáu chất chuyển hóa chính của Ethionamide đã được phát hiện bao gồm 2-ethylisonicotinamide, carbonyl-dihydropyridine, thiocarbonyl-dihydropyridine, S-oxocarbamoyl dihydropyridine, 2-ethylthioiso-nicotinamide và ethionamide sulfoxide. Chất chuyển hóa có hoạt tính là dẫn chất sulfoxide. Prothionamide cũng được chuyển hóa tại gan thành các chất chuyển hóa hoạt động (dẫn chất sulfoxide) và không hoạt động.
- Thải trừ: t1/2 của Ethionamide khoảng 1.92 giờ. Chỉ có không quá 1% liều Ethionamide và Prothionamide được bài xuất dưới dạng không đổi qua nước tiểu.
Cycloserine
- Hấp thu: Hấp thu nhanh và gần như hoàn toàn sau khi uống. Tmax khoảng 3-4 giờ.
- Phân bố: Phân bố tốt vào các mô và dịch trong cơ thể. Nồng độ thuốc trong dịch não tủy gần ngang với trong huyết tương. Thuốc cũng vào được đờm, dịch màng phổi, dịch cổ trướng, mật, nước ối, máu thai nhi và sữa mẹ.
- Chuyển hóa: Có khoảng 35% liều được chuyển hóa, nhưng không rõ chất chuyển hóa là gì.
- Thải trừ: Thuốc được bài tiết vào nước tiểu. Khoảng 66% liều xuất hiện trong nước tiểu dưới dạng không đổi trong vòng 24 giờ. 10% liều tiếp tục xuất hiện trong nước tiểu trong vòng 48 giờ tiếp theo. Bài xuất qua phân không đáng kể. t1/2 khoảng từ 8-12 giờ.
Para-aminosalicylic acid
- Hấp thu: Chế phẩm chứa Para-aminosalicylic acid được bào chế dưới dạng viên bao tan trong ruột (kháng acid dạ dày). Điều này giúp cản trở hình thành chất gây độc gan m-aminophenol (độc với gan). Tmax khoảng 6 giờ.
- Phân bố: Phân bố tốt vào các mô và dịch trong cơ thể, bao gồm gan, thận, phổi… Thuốc chỉ qua được hàng rào máu não khi màng não bị viêm. Không rõ thuốc có qua được hàng rào nhau thai hay không. Liên kết với protein huyết tương khoảng 50-60%. Thể tích phân bố (Vd) khoảng 1.001 L/kg.
- Chuyển hóa: Thuốc được acetyl hóa tại gan và chất chuyển hóa tạo thành là N-acetyl-p-aminosalicylic acid không có hoạt tính. Ngoài ra, một chất chuyển hóa khác là p-aminosalicyluric acid được liên hợp với glycine.
- Thải trừ: t1/2 trong huyết tương khoảng 1-2 giờ. Thải trừ thuốc chủ yếu qua nước tiểu thông qua lọc ở cầu thận và bài tiết ở ống thận.
Clofazimine
- Hấp thu: Hấp thu theo đường uống thay đổi, với hỗn dịch sáp – dầu là 70%. Dùng thuốc cùng với thức ăn làm tăng Cmax (nồng độ đỉnh trong huyết tương) nhưng kéo dài Tmax.
- Phân bố: Phân bố rộng vào các mô trong cơ thể. Thuốc không qua được hàng rào máu não, nhưng qua được nhau thai và sữa mẹ.
- Chuyển hóa: Chuyển hóa thuốc diễn ra tại gan. Cho đến nay đã biết ba chất chuyển hóa của Clofazimine xuất hiện trong nước tiểu.
- Thải trừ: Thuốc được bài xuất qua cả phân và nước tiểu. t1/2 có thể lên đến 70 ngày.
Terizidone
Dữ liệu về dược động học của Terizidone trong tài liệu rất hạn chế.
- Hấp thu: Hấp thu tốt qua đường tiêu hóa. Tmax khoảng 3 giờ. Hằng số hấp thu ka = 1.17-1.36 h-1.
- Phân bố: Thế tích phân bố cao, dao động trong khoảng 113-246 L.
- Chuyển hóa: Terizidone được chuyển hóa thành Cycloserine trong cơ thể, có lẽ tại gan, nhưng cách thức chuyển hóa nó chưa rõ.
- Thải trừ: 39% liều được bài xuất qua nước tiểu trong vòng 30 giờ.
Thiacetazone
Dữ liệu về dược động học của Thiacetazone trong tài liệu cũng rất hạn chế. Dữ liệu dưới đây lấy từ một nghiên cứu lâm sàng pha I, nhãn mở trên 12 người khỏe mạnh với liều Thiacetazone 150 mg/ngày trong 7 ngày.
- Hấp thu: Cmax = 1.59 ± 0.47 µg/mL, Tmax = 3.30 ± 1.18 giờ.
- Phân bố: Không rõ.
- Chuyển hóa: Không rõ.
- Thải trừ: t1/2 = 15-16 giờ. Không quá 25% liều Thiacetazone được thu hồi dưới dạng không đổi trong nước tiểu trong vòng 48 giờ.
Thioridazine
- Hấp thu: Sinh khả dụng của thuốc khoảng 60%.
- Phân bố: Liên kết với protein huyết tương là 95%.
- Chuyển hóa: Thuốc được chuyển hóa tại gan. Các chất chuyển hóa là Thioridazine 2-sulfoxide, N-desmethylthioridazine và Thioridazine 5-sulfoxide.
- Thải trừ: t1/2 = 21-25 giờ.
Bedaquiline
- Hấp thu: Tmax khoảng 5 giờ. Dùng cùng bữa ăn giàu chất béo làm tăng sinh khả dụng của thuốc.
- Phân bố: Liên kết với protein huyết tương cao, trên 99.9%. Vd trong ngăn trung tâm xấp xỉ 164 L.
- Chuyển hóa: Thuốc được chuyển hóa bởi CYP3A4 ở gan, chất chuyển hóa chính là M2, dẫn xuất N-monodesmethyl.
- Thải trừ: t1/2 trung bình của Bedaquiline và chất chuyển hóa M2 là 5.5 tháng. Thải trừ thuốc chủ yếu qua phân.
Delamanid và Pretomanid
- Hấp thu: Tmax của Delamanid khoảng 4-5 giờ. Nồng độ Delamanid trong máu ổn định sau 10-14 ngày dùng thuốc. Sinh khả dụng ước tính của Delamanid là 25-47%. Dùng cả hai thuốc cùng thức ăn giàu chất béo làm tăng sinh khả dụng của thuốc.
- Phân bố: Vd biểu kiến của Delamanid là 2100 L. Cả hai thuốc có thể vào được sữa mẹ (theo các nghiên cứu trên động vật). Liên kết với protein huyết tương của Delamanid là trên 99.5% và của Pretomanid và 86.4%.
- Chuyển hóa: Delamanid và Pretomanid được chuyển hóa ở gan. Bốn chất chuyển hóa chính của Delamanid là M1, M2, M3 và M4, trong đó M1 góp phần chủ yếu vào tác dụng phụ gây kéo dài khoảng QT của thuốc. Pretomanid được chuyển hóa theo các con đường khử và oxy hóa, nhưng không có con đường chính. CYP3A ở gan chỉ chịu trách nhiệm chuyển hóa 20% Pretomanid.
- Thải trừ: t1/2 của Delamanid là 30-38 giờ sau khi ngừng thuốc. Delamanid được bài xuất chủ yếu qua phân, dưới 5% được bài xuất qua nước tiểu. Trong khi đó, Pretomanid được bài xuất qua nước tiểu nhiều hơn.
Tác dụng – Chỉ định
Các thuốc trong nhóm này chủ yếu được sử dụng trong điều trị bệnh lao. Ngoài ra, một số thuốc có thể có thêm những chỉ định khác, như Ethionamide, Prothionamide, Clofazimine và Rifampicin có thể được chỉ định trong bệnh phong. Bản thân Rifampicin cũng được chỉ định trong một số nhiễm trùng nghiêm trọng do một số vi khuẩn kháng thuốc gây ra.
Isoniazid, Rifampicin, Pyrazinamide và Ethambutol là các thuốc điều trị lao đầu tay, và đôi khi có cả Streptomycin.

Các thuốc còn lại chủ yếu sử dụng tuyến hai, tức là khi phác đồ đầu tay không mang lại hiệu quả do vi khuẩn lao kháng thuốc.
Cách dùng – Liều dùng
- Isoniazid: 4-5 mg/kg/ngày. Tối đa có thể lên đến 300 mg/ngày. Ở bệnh nhân mắc lao màng não, liều dùng trong 1-2 tuần đầu có thể lên tới 10 mg/kg/ngày. Một số phác đồ khác có thể sử dụng liều Isoniazid 15 mg/kg 2-3 lần/tuần. Isoniazid nên được dùng khi dạ dày rỗng, tốt nhất là 30 phút trước bữa ăn.
- Rifampicin: 8-12 mg/kg/ngày. Thông thường, người có cân nặng dưới 50 kg dùng liều 450 mg/ngày, người có cân nặng từ 50 kg trở lên dùng liều 600 mg/ngày.
- Pyrazinamide: Người nặng dưới 50 kg, dùng tối đa 1.5 g/ngày hoặc 2 g 3 lần/tuần, người nặng trên 50 kg, dùng tối đa 2 g/ngày hoặc 2.5 g 3 lần/tuần.
- Ethambutol: 15-25 mg/kg/ngày.
- Capreomycin: Tiêm bắp sâu hoặc truyền tĩnh mạch 1 g/ngày (không quá 20 mg/kg/ngày) trong 60-120 ngày, sau đó dùng thuốc liều 1 g 2-3 lần/tuần.
- Ethionamide: 15-20 mg/kg/ngày, tối đa 1 g/ngày.
- Prothionamide: 15-20 mg/kg/ngày, tối đa 1 g/ngày. Với trẻ sơ sinh, trẻ em và trẻ vị thành niên: 7.5-15 mg/kg/ngày, tối đa 500 mg/ngày.
- Cycloserine: 250 mg mỗi 12 giờ trong 2 tuần đầu tiên. Liều hàng ngày không vượt quá 1 g.
- Para-aminosalicylic acid: 4 g 3 lần/ngày. Liều tối đa 12 g/ngày.
- Clofazimine: 100 mg 1-3 lần/ngày.
- Terizidone, Thiacetazone: Các thuốc này không còn được lưu hành tại châu Âu, Hoa Kỳ hay Việt Nam.
- Thioridazine: Thuốc vẫn được lưu hành nhưng không còn được sử dụng cho chỉ định điều trị lao, mà chủ yếu dùng trong điều trị tâm thần phân liệt và rối loạn trầm cảm.
- Bedaquiline: 400 mg/ngày trong 2 tuần đầu, sau đó 200 mg 3 lần/tuần, khoảng cách giữa các liều cách nhau ít nhất 48 giờ.
- Delamanid: 100 mg 2 lần/ngày, uống cùng thức ăn. Trẻ em và trẻ vị thành niên có cân nặng dưới 50 kg nhưng từ 30 kg trở lên dùng liều 50 mg 2 lần/ngày.
- Pretomanid: 200 mg/ngày.
Tác dụng không mong muốn
- Isoniazid: Tác dụng không mong muốn nổi bật của thuốc là độc tính trên gan và gây thiếu vitamin B6 (do cấu trúc tương tự vitamin B6), dẫn đến một loạt các bệnh lý thần kinh ngoại biên. Một số tác dụng không mong muốn khác là rối loạn tiêu hóa (táo bón, khô miệng, buồn nôn), mất bạch cầu hạt, thiếu máu bất sản, điếc, ù tai, rối loạn tâm thần, lupus ban đỏ hệ thống hoặc hội chứng giống lupus…
- Rifampicin: Thuốc cũng có độc tính trên gan, nhưng nhẹ hơn Isoniazid. Ngoài ra, trong quá trình sử dụng thuốc, các dịch trên cơ thể có thể chuyển sang màu đỏ hoặc cam. Đây là phản ứng hết sức bình thường của thuốc và nó không gây nguy hiểm. Một số tác dụng không mong muốn khác là rối loạn tiêu hóa (giảm thèm ăn, nôn, buồn nôn, tiêu chảy, đổi màu răng, viêm đại tràng giả mạc…), dị ứng (có thể nhẹ như phát ban, ngứa, cũng có thể nặng như hội chứng Stevens-Johnson, hội chứng Lyell, hội chứng ngoại ban mụn mủ toàn thân cấp AGEP…), đau đầu, chóng mặt, rối loạn đông máu phụ thuộc vitamin K, đông máu nội mạch rải rác (DIC), giảm bạch cầu, giảm tiểu cầu…
- Pyrazinamide: Thuốc cũng có độc tính trên gan, nhưng nhẹ hơn Isoniazid. Một số tác dụng không mong muốn khác là rối loạn tiêu hóa (chán ăn, buồn nôn và nôn), sốt, phát ban, mày đay, ngứa…
- Ethambutol: Viêm dây thần kinh sau nhãn cầu, đi kèm với giảm thị lực là tác dụng không mong muốn phổ biến nhất. Một số tác dụng không mong muốn khác là rối loạn tiêu hóa (nôn, buồn nôn, tiêu chảy…), đau khớp, phát ban, ngứa, viên thận kẽ, viêm phổi, viêm dây thần kinh ngoại biên, tăng acid uric máu, gout, giảm bạch cầu, giảm tiểu cầu…
- Capreomycin: Thuốc có độc tính trên thận và tai, gây viêm thận, suy thận, ù tai, chóng mặt. Rối loạn điện giải, bao gồm hạ kali máu, hạ magnesi máu và hạ calcium máu có thể xảy ra. Bạch cầu có thể tăng hoặc giảm. Ngoài ra, bệnh nhân có thể bị đau tại nơi tiêm, áp xe vô trùng, sốt, mày đay, phát ban…
- Ethionamide và Prothionamide: Rối loạn tiêu hóa (buồn nôn, nôn, tiêu chảy, đau bụng, lưỡi vị kim loại…), rối loạn tâm thần, buồn ngủ, chóng mặt, nhức đầu, viêm dây thần kinh ngoại biên, viêm dây thần kinh thị giác, viêm gan, phát ban, nhạy cảm ánh sáng hoặc các phản ứng dị ứng da nghiêm trọng. Một số tác dụng không mong muốn khác là hạ đường huyết, nữ hóa tuyến vú ở nam giới, liệt dương…
- Cycloserine: Phổ biến nhất là tác dụng không mong muốn trên hệ thần kinh, bao gồm co giật, buồn ngủ, đau đầu, chóng mặt, lú lẫn, mất phương hướng, rối loạn tâm thần, dị cảm, liệt dương… Suy tim sung huyết cũng được báo cáo ở những bệnh nhân dùng liều 1-1.5 g/ngày. Một số tác dụng không mong muốn khác là dị ứng, phát ban da, tăng men gan…
- Para-aminosalicylic acid: Hầu hết các tác dụng không mong muốn liên quan đến hệ tiêu hóa như buồn nôn, nôn, chướng bụng, tiêu chảy, chán ăn… Các tác dụng không mong muốn khác có thể liên quan đến thần kinh như chóng mặt hội chứng tiền đình, thị giác bất thường, bệnh lý thần kinh ngoại biên… hoặc dị ứng trên da như quá mẫn, phát ban, ngứa…
- Clofazimine: Rối loạn tiêu hóa như buồn nôn, nôn, tiêu chảy, đau bụng… Đổi màu nước tiểu, phân và da. Có thể gặp dị ứng da như ngứa, phát ban…
- Bedaquiline: Buồn nôn, đau khớp, đau đầu và đau ngực là các tác dụng không mong muốn phổ biến nhất. Các tác dụng không mong muốn ít phổ biến hơn là chán ăn, phát ban, tăng men gan và kéo dài khoảng QT.
- Delamanid: Các tác dụng không mong muốn phổ biến nhất là buồn nôn, nôn, đau đầu, mất ngủ, chóng mặt, ù tai, hạ kali máu, viêm dạ dày, chán ăn và suy nhược.
- Pretomanid: Thuốc có độc tính với gan. Đây là tác dụng không mong muốn đặc trưng của thuốc. Ngoài ra, các tác dụng không mong muốn khác có thể xảy ra là rối loạn tiêu hóa, dị ứng trên da…
Chống chỉ định
Quá mẫn cảm với bất cứ thành phần nào của thuốc.
- Isoniazid: Bệnh gan do thuốc.
- Rifampicin: Vàng da, hoặc đang sử dụng đồng thời Saquinavir/Ritonavir.
- Pyrazinamide: Bệnh gan, tăng acid uric máu, bệnh gout, rối loạn chuyển hóa porphyrin cấp.
- Ethambutol: Viêm dây thần kinh thị giác và thị lực kém hoặc viêm dây thần kinh sau nhãn cầu (trừ khi lợi ích là vượt trội so với nguy cơ).
- Ethionamide: Bệnh nhân suy gan nặng, tiền sử dị ứng nặng với Prothionamide.
- Prothionamide: Bệnh nhân suy gan nặng, co giật do não, rối loạn tâm thần, tiền sử dị ứng nặng với Ethionamide.
- Cycloserine: Động kinh, trầm cảm, rối loạn tâm thần, lo lắng quá mức, suy thận nặng, sử dụng quá nhiều rượu đồng thời.
- Para-aminosalicylic acid: Suy thận nặng.
- Delamanid: Nồng độ albumin huyết thanh dưới 2.8 g/dL hoặc phối hợp với các thuốc cảm ứng CYP3A4 của gan mạnh.
Tương tác thuốc
Isoniazid:
- Phối hợp với các thuốc độc với gan khác (đặc biệt là các thuốc cùng phác đồ điều trị lao): Tăng nguy cơ gặp phải độc tính trên gan.
- Isoniazid ức chế chuyển hóa một số thuốc dùng cùng và làm tăng độc tính của chúng, đặc biệt là Carbamazepine, Primidone, Phenytoin, Diazepam và Triazolam.
- Isoniazid ức chế monoamine oxidase (MAO) và diamine oxidase (DAO), gây giảm chuyển hóa tyramine và histamine. Khuyến cáo không nên sử dụng các thực phẩm giàu tyramine và histamine (thịt đã qua chế biến, rượu vang, bia, cá thu, cá hồi, cá ngừ…) trong quá trình điều trị bằng Isoniazid do có thể gây ra các phản ứng đánh trống ngực, đổ mồ hôi, hạ huyết áp…
- Phối hợp với Itraconazole: Nồng độ Itraconazole trong máu giảm đáng kể và có thể dẫn đến thất bại điều trị. Không nên sử dụng phối hợp này.
- Phối hợp với Zalcitabine (điều trị HIV/AIDS): Tăng thanh thải của Isoniazid, có thể dẫn đến giảm tác dụng điều trị lao. Cần cân nhắc tăng liều Isoniazid trong trường hợp này.
Rifampicin:
- Phối hợp với các thuốc độc với gan khác (đặc biệt là các thuốc cùng phác đồ điều trị lao): Tăng nguy cơ gặp phải độc tính trên gan. Tránh phối hợp Rifampicin với Halothane.
- Phối hợp với Saquinavir/Ritonavir: Chống chỉ định. Các thuốc dùng cùng này ức chế CYP3A4 của gan mạnh và làm tăng nồng độ Rifampicin trong máu, tăng nguy cơ nhiễm độc gan.
- Phối hợp với các kháng sinh khác có thể gây rối loạn đông máu phụ thuộc vitamin K (Cefazolin, các kháng sinh Cephalosporin khác có chuỗi N-methyl-thiotetrazole): Tránh phối hợp này do làm nặng hơn tình trạng rối loạn đông máu của bệnh nhân.
- Rifampicin là chất cảm ứng CYP 1A2, 2B6, 2C8, 2C9, 2C19 và 3A4, cảm ứng UDP-glucuronyltransferase (UGT), sulfotransferase, carboxylesterase, P-gp (P-glycoprotein), MRP2 (protein liên quan đến kháng đa thuốc). Do đó các thuốc là cơ chất của những enzyme hoặc protein vận chuyển này khi dùng cùng Rifampicin sẽ bị giảm nồng độ trong máu, tăng nguy cơ thất bại điều trị. Các thuốc bị ảnh hưởng có thể là: Quinidine, Phenytoin, Tamoxifen, Haloperidol, Fluconazole, Itraconazole, Saquinavir, Lopinavir, Bisoprolol, Diazepam, Zolpidem, Diltiazem, Nifedipine, Verapamil, Clarithromycin, Dapsone, các corticoid, sulfonylurea điều trị đái tháo đường, Cyclosporin, Tacrolimus, Losartan, Enalapril, các thuốc điều trị viêm gan C…
- Phối hợp với thuốc kháng acid dạ dày: Hấp thu Rifampicin giảm. Nên dùng Rifampicin ít nhất 1 giờ trước khi dùng thuốc kháng acid.
Pyrazinamide:
- Phối hợp với các thuốc độc với gan khác (đặc biệt là các thuốc cùng phác đồ điều trị lao): Tăng nguy cơ gặp phải độc tính trên gan.
- Đối kháng tác dụng với Probenecid và Sulfinpyrazone.
- Phối hợp với thuốc tránh thai đường uống chứa Estrogen: Giảm tác dụng tránh thai.
- Phối hợp với vaccin thương hàn: Tác dụng của vaccin giảm. Không nên dùng thuốc trong vòng 3 ngày trước và sau khi tiêm vaccin.
Ethambutol:
- Phối hợp với chế phẩm chứa nhôm hydroxide: Hấp thu Ethambutol giảm. Nên dùng thuốc kháng acid dạ dày không chứa nhôm hydroxide khi đang điều trị bằng Ethambutol.
Capreomycin:
- Phối hợp với các thuốc khác có độc tính trên thận hoặc thính giác: Hết sức thận trọng do làm tăng độc tính trên hai cơ quan này.
Ethionamide và Prothionamide:
- Phối hợp với Cycloserine có báo cáo co giật.
- Sử dụng quá nhiều Ethanol trong quá trình điều trị có thể dẫn đến loạn thần.
Cycloserine:
- Phối hợp với Ethionamide hoặc Prothionamide làm tăng độc tính thần kinh (có thể gây co giật).
- Phối hợp với Ethanol: Bệnh nhân uống nhiều rượu trong quá trình điều trị làm tăng nguy cơ xuất hiện động kinh.
- Phối hợp với Isoniazid làm tăng nguy cơ gặp tác dụng không mong muốn trên thần kinh trung ương.
Para-aminosalicylic acid:
- Thuốc có thể gây giảm hấp thu vitamin B12, gây ra bất thường hồng cầu có ý nghĩa trên lâm sàng. Bệnh nhân điều trị bằng thuốc này từ một tháng trở lên nên được bổ sung vitamin B12.
- Phối hợp với Digoxin: Hấp thu Digoxin giảm do Para-aminosalicylic acid ức chế chức năng hấp thu Digoxin của niêm mạc ruột.
- Phối hợp với Ethionamide: Tăng các tác dụng không mong muốn trên hệ tiêu hóa của Para-aminosalicylic acid.
- Phối hợp với Diphenylhydramine: Hấp thu Para-aminosalicylic acid bị giảm. Không nên dùng hai thuốc này đồng thời.
Clofazimine:
- Phối hợp với các thuốc có thể gây kéo dài khoảng QT: Tăng nguy cơ gặp phải kéo dài khoảng QT trên điện tâm đồ. Thận trọng với phối hợp này, đặc biệt nên tránh phối hợp với các thuốc Entrectinib, Glasdegib, Hydroxychloroquine, Ivosidenib, Lefamulin, Macimorelin, Pitolisant và Ribociclib.
Bedaquiline:
- Phối hợp với các thuốc có thể gây kéo dài khoảng QT: Tăng nguy cơ gặp phải kéo dài khoảng QT trên điện tâm đồ. Thận trọng với phối hợp này. Đặc biệt chống chỉ định phối hợp Bedaquiline với Lefamulin.
- Phối hợp với các thuốc cảm ứng hoặc ức chế enzyme CYP3A4 ở gan: Nồng độ Bedaquiline trong máu có thể giảm hoặc tăng tương ứng, dẫn đến nguy cơ chọn lọc các chủng vi khuẩn lao đề kháng thuốc hoặc nguy cơ gặp phải độc tính của thuốc, tương ứng. Đặc biệt nên tránh phối hợp Bedaquiline với các thuốc ức chế protease HIV, kháng nấm azole, sulfonylurea điều trị đái tháo đường, Carbamazepine, Phenytoin, các barbiturate, Efavirenz…
- Vaccin BCG làm giảm tác dụng của Bedaquiline. Nên tránh phối hợp này.
Delamanid:
- Phối hợp với các thuốc cảm ứng hoặc ức chế enzyme CYP3A4 ở gan: Tương tự như Bedaquiline. Đặc biệt phối hợp với thuốc cảm ứng CYP3A4 mạnh như Carbamazepine bị chống chỉ định.
Pretomanid:
- Phối hợp với rượu bia hoặc các thuốc gây độc cho gan: Tăng nguy cơ gặp phải độc tính trên gan.
- Phối hợp với thuốc cảm ứng CYP3A4 mạnh như Rifampicin hoặc Efavirenz: Nên tránh phối hợp này do nồng độ thuốc trong máu có thể giảm đáng kể.
Lưu ý và thận trọng khi sử dụng thuốc
Isoniazid: Tất cả bệnh nhân cần được xét nghiệm chức năng gan trước và trong quá trình điều trị thường xuyên. Bác sĩ sẽ xem xét quyết định trường hợp nào cần ngừng thuốc. Thận trọng khi sử dụng Isoniazid cho các bệnh nhân đái tháo đường, rối loạn chức năng gan, nghiện rượu lâu năm, tiền sử rối loạn tâm thần, bệnh nhân có HIV dương tính, lao ngoài phổi, hoặc có kiểu hình acetyl hóa chậm… Ngoài ra, cần xem xét bổ sung vitamin B6 trong quá trình điều trị.
Rifampicin: Tất cả bệnh nhân cần được xét nghiệm chức năng gan trước và trong quá trình điều trị thường xuyên. Bác sĩ sẽ xem xét quyết định trường hợp nào cần ngừng thuốc. Theo dõi thận trọng các phản ứng trên da. Cân nhắc dùng vitamin K cho bệnh nhân để tránh rối loạn đông máu nghiêm trọng phụ thuộc vitamin K.
Pyrazinamide: Tất cả bệnh nhân cần được xét nghiệm chức năng gan trước và trong quá trình điều trị thường xuyên. Bác sĩ sẽ xem xét quyết định trường hợp nào cần ngừng thuốc. Ngoài chức năng gan, nồng độ acid uric trong máu cũng cần được định lượng thường xuyên. Thận trọng với bệnh nhân suy thận, có thể cần giảm liều hoặc tần suất dùng thuốc. Thận trọng khi sử dụng thuốc cho phụ nữ có thai, bệnh nhân có tiền sử gout hoặc đái tháo đường.
Ethambutol: Bệnh nhân cần báo cáo bất cứ thay đổi nào nếu thấy thị lực giảm khi đang điều trị bằng thuốc. Thị lực thường có thể hồi phục trong vòng vài tuần nếu ngừng thuốc ngay lập tức. Kiểm tra chức năng thận của bệnh nhân trước khi điều trị để có chế độ liều thích hợp. Xét nghiệm chức năng gan nếu bệnh nhân có gợi ý cho thấy viêm gan.
Capreomycin: Thận trọng khi sử dụng trên nền bệnh nhân suy thận hoặc suy giảm thính lực. Đánh giá chức năng thính lực, tiền đình và chức năng thận nên được tiến hành thường xuyên. Không khuyến cáo phối hợp với Streptomycin.
Ethionamide và Prothionamide:
- Theo dõi chức năng gan trước và trong quá trình điều trị thường xuyên. Bác sĩ sẽ xem xét quyết định trường hợp nào cần ngừng thuốc.
- Kiểm tra định kỳ chức năng tuyến giáp, thị giác và đường huyết.
- Thận trọng với bệnh nhân có rối loạn tâm thần, trầm cảm, suy thận, loét dạ dày – tá tràng, ho ra máu, rối loạn đông máu.
- Các phản ứng có hại trên da nghiêm trọng như hội chứng Stevens-Johnson, hoại tử thượng bì nhiễm độc, phản ứng thuốc với tăng bạch cầu ái toan và triệu chứng toàn thân DRESS, ngoại ban mụn mủ toàn thân cấp AGEP… đã được báo cáo với Ethionamide. Theo dõi nếu bệnh nhân có phát ban trên da và xem xét ngừng thuốc nếu các triệu chứng tiến triển xấu.
- Bệnh nhân nên được sử dụng đồng thời với vitamin B6.
Cycloserine:
- Ngừng sử dụng hoặc giảm liều thuốc nếu bệnh nhân gặp phải các triệu chứng của viêm da dị ứng hoặc độc tính thần kinh trung ương như co giật, rối loạn tâm thần, trầm cảm…
- Nguy cơ co giật ở bệnh nhân nghiện rượu tăng lên.
- Theo dõi các dấu hiệu huyết học, chức năng gan trong quá trình điều trị.
Para-aminosalicylic acid: Thận trọng ở những bệnh nhân suy thận từ nhẹ đến trung bình, suy gan, loét dạ dày – tá tràng. Ngừng sử dụng thuốc nếu bệnh nhân có biểu hiện viêm gan, phát ban, sốt, không dung nạp thuốc trong quá trình điều trị. Para-aminosalicylic acid còn có thể làm tăng nguy cơ suy giáp ở bệnh nhân có lao đồng nhiễm HIV. Ở những bệnh nhân này, chức năng tuyến giáp cần được theo dõi trước điều trị và thường xuyên trong quá trình điều trị, đặc biệt khi thuốc được sử dụng cùng với Ethionamide hoặc Prothionamide.
Clofazimine:
- Tư vấn cho bệnh nhân về sự thay đổi màu da cũng như một số bộ phận khác trong quá trình dùng thuốc. Theo dõi trầm cảm.
- Thận trọng với các tác dụng không mong muốn trên đường tiêu hóa. Liều trên 100 mg chỉ nên sử dụng trong thời gian ngắn (dưới ba tháng). Các triệu chứng tiêu hóa nghiêm trọng có thể đòi hỏi giảm liều, giãn liều hoặc ngừng thuốc.
- Kéo dài khoảng QT có thể xảy ra, đặc biệt ở những bệnh nhân dùng liều trên 100 mg/ngày. Theo dõi điện tâm đồ thường xuyên trong trường hợp này.
Bedaquiline:
- Rất thận trọng khi phối hợp thuốc này với các thuốc khác cũng có khả năng gây kéo dài khoảng QT, hoặc bệnh nhân có tiền sử kéo dài khoảng QT.
- Đã có một thử nghiệm lâm sàng cho thấy tỷ lệ tử vong ở nhóm dùng Bedaquiline cao hơn so với nhóm dùng giả dược (11.4% vs. 2.5%).
- Chỉ sử dụng khi các phác đồ điều trị lao trước đó không còn hiệu quả.
- Theo dõi các tác dụng không mong muốn trên gan. Trong quá trình điều trị, cố gắng tránh uống rượu bia hoặc các thuốc gây độc gan khác, đặc biệt trên nền bệnh nhân suy gan.
- Delamanid: Không khuyến cáo sử dụng thuốc cho bệnh nhân suy gan từ trung bình đến nặng.
- Pretomanid: Tránh sử dụng rượu hoặc các thuốc có độc tính trên gan trong quá trình điều trị. Bác sĩ có thể quyết định ngừng thuốc nếu phát hiện ra một số bất thường đáng kể.
Sử dụng thuốc trên đối tượng đặc biệt
Isoniazid:
- Chỉ sử dụng thuốc cho phụ nữ mang thai khi lợi ích nhận được là vượt trội so với nguy cơ có thể gặp phải trên thai nhi. Tuy nhiên, thông thường thì khi mắc bệnh lao, lợi ích nhận được từ việc điều trị sẽ luôn lớn hơn. Với phụ nữ đang cho con bú, theo dõi các dấu hiệu ngộ độc có thể xảy ra ở trẻ sơ sinh.
- Thận trọng khi sử dụng thuốc ở người già do chức năng gan và thận có thể suy giảm.
- Không sử dụng thuốc cho trẻ 0-3 tháng tuổi do thiếu dữ liệu lâm sàng.
Rifampicin:
- Tác dụng trên bào thai ở người chưa rõ. Chỉ sử dụng thuốc cho phụ nữ mang thai khi lợi ích nhận được là vượt trội so với nguy cơ có thể gặp phải trên thai nhi. Nếu Rifampicin được sử dụng trong những tuần cuối thai kỳ, thuốc có thể gây xuất huyết sau sinh cho cả mẹ và trẻ, điều trị bằng vitamin K1.
- Không nên cho con bú khi đang sử dụng thuốc. Thông thường bệnh nhân sẽ được yêu cầu cho con dùng sữa ngoài.
Pyrazinamide:

- Chỉ sử dụng thuốc cho phụ nữ mang thai khi lợi ích nhận được là vượt trội so với nguy cơ có thể gặp phải trên thai nhi.
- Với phụ nữ đang cho con bú, cần cho trẻ dùng sữa ngoài trong thời gian dùng thuốc.
Ethambutol:
- Chỉ sử dụng thuốc cho phụ nữ mang thai khi lợi ích nhận được là vượt trội so với nguy cơ có thể gặp phải trên thai nhi.
- Với phụ nữ đang cho con bú, nên cho trẻ dùng sữa ngoài trong thời gian dùng thuốc.
Capreomycin:
- Chỉ sử dụng thuốc cho phụ nữ mang thai khi lợi ích nhận được là vượt trội so với nguy cơ có thể gặp phải trên thai nhi.
- Thận trọng khi sử dụng thuốc cho phụ nữ đang cho con bú (không rõ thuốc có vào được sữa mẹ hay không).
Ethionamide và Prothionamide:
- Chỉ sử dụng thuốc cho phụ nữ mang thai khi lợi ích nhận được là vượt trội so với nguy cơ có thể gặp phải trên thai nhi.
- Chỉ sử dụng thuốc cho phụ nữ đang cho con bú nếu lợi ích nhận được là vượt trội so với nguy cơ cho trẻ bú mẹ. Nếu có thể, nên cho trẻ dùng sữa ngoài trong quá trình điều trị cho người mẹ.
Cycloserine:
- Phân loại an toàn trong thai kỳ: C. Chỉ sử dụng thuốc cho phụ nữ có thai khi thực sự cần thiết.
- Với phụ nữ đang cho con bú, nên cho con sử dụng sữa ngoài trong quá trình điều trị.
Para-aminosalicylic acid:
- Không khuyến cáo sử dụng thuốc cho phụ nữ có thai. Chỉ sử dụng thuốc khi thực sự cần thiết.
- Với phụ nữ đang cho con bú, nên cho con sử dụng sữa ngoài trong quá trình điều trị.
Clofazimine:
- Phân loại an toàn trong thai kỳ: C. Chỉ sử dụng thuốc cho phụ nữ có thai khi thực sự cần thiết.
- Không nên dùng thuốc cho phụ nữ đang cho con bú. Nếu bắt buộc phải sử dụng, nên cho trẻ dùng sữa ngoài.
Bedaquiline:
- Dữ liệu lâm sàng trên phụ nữ mang thai còn thiếu. Chỉ sử dụng thuốc cho phụ nữ có thai khi thực sự cần thiết.
- Không có dữ liệu về sự hiện diện của Bedaquiline trong sữa mẹ. Nếu có thể, tốt nhất nên cho trẻ dùng sữa ngoài trong thời gian người mẹ điều trị bằng Bedaquiline.
Delamanid và Pretomanid:
- Không khuyến khích sử dụng cho phụ nữ có thai do dữ liệu hạn chế. Chỉ sử dụng thuốc trên những đối tượng này khi thực sự cần thiết.
- Không có dữ liệu về sự hiện diện của Delamanid trong sữa mẹ. Nếu bắt buộc người mẹ phải sử dụng thuốc trong thời kỳ cho con bú, nên cho trẻ dùng sữa ngoài.
Một số nghiên cứu và thử nghiệm lâm sàng
Đánh giá hệ thống so sánh Bedaquiline với giả dược trong điều trị bệnh lao kháng đa thuốc đưa ra kết luận: Bedaquiline hiệu quả trong điều trị lao kháng đa thuốc, tuy nhiên cần phải cân bằng lợi ích này với tỷ lệ tử vong cao đáng kể. Cần phải có thêm nhiều thử nghiệm lâm sàng pha III hơn trong tương lai để có thể đánh giá đúng vai trò của Bedaquiline để đưa ra khuyến nghị điều trị chính xác.
Thử nghiệm Nix-TB, một nghiên cứu nhãn mở đánh giá hiệu lực và an toàn của phác đồ chứa ba thuốc Bedaquiline, Pretomanid và Linezolid trong điều trị lao kháng thuốc rộng hoặc lao kháng đa thuốc không dung nạp/không đáp ứng trong thời gian 26 tuần cho kết luận: Phác đồ chứa ba thuốc này là khả thi cho các bệnh nhân có các dạng lao kháng thuốc cao.
Kháng sinh điều trị phong
Giới thiệu chung
Lịch sử ra đời
Giống với bệnh lao, phong cũng từng là một trong “tứ chứng nan y”. Trước đây, phong là một bệnh không thể chữa khỏi, bệnh nhân mắc bệnh phong không chỉ phải chịu nỗi đau về thể xác mà còn phải chịu nỗi đau về tinh thần rất lớn. Họ thường bị kì thị, khinh thường và cô lập với xã hội. Tuy vậy, với trình độ phát triển của y học hiện đại, phong hiện nay đã là một bệnh có thể được điều trị khỏi hoàn toàn nếu được phát hiện sớm và điều trị kịp thời.
Phong là một bệnh lý nhiễm trùng do vi khuẩn M. leprae gây ra. Vi khuẩn này cùng thuộc chi Mycobacterium như trực khuẩn lao và cũng là một trực khuẩn kháng cồn – acid. Không giống với lao, phong là một bệnh rất khó lây. Có nhiều nguyên nhân có thể lý giải cho điều này. Thứ nhất, phải tiếp xúc với người bệnh đủ lâu trong một thời gian dài mới có thể truyền bệnh. Thứ hai, nhiều người có miễn dịch với vi khuẩn (khoảng 95% dân số có miễn dịch với bệnh phong). Thứ ba, chỉ có hai thể phong là B và L mang nhiều vi khuẩn mới có khả năng truyền bệnh đáng kể. Thứ tư, chu kỳ phát triển và sinh sản của trực khuẩn phong rất chậm, nên nó thường bị hệ miễn dịch của cơ thể tiêu diệt trước khi có khả năng lây bệnh. Và cuối cùng, với sự hiệu quả của các phác đồ điều trị phong hiện nay, phong gần như là một bệnh không thể lây truyền nếu được điều trị đúng.
Không giống như lao, phong ít kháng thuốc hơn. Từ năm 1981 đến nay, phác đồ điều trị phong tiêu chuẩn của Tổ chức Y tế Thế giới (WHO) bao gồm ba thuốc: Dapsone, Clofazimine và Rifampicin. Các thuốc khác có thể được sử dụng thay thế khi các thuốc đầu tay không hiệu quả do vi khuẩn kháng thuốc hoặc bệnh nhân không dung nạp là Ethionamide, Prothionamide, Thalidomide hoặc Sulfoxone. Các thuốc đã được trình bày trong phần A sẽ ít được nhắc lại trong phần này.
Dapsone là thuốc điều trị bệnh phong được nghiên cứu lâu đời nhất và vẫn là một trong các thuốc hiệu quả nhất cho đến nay. Thuốc được nghiên cứu từ năm 1937 và đến năm 1945 đã bắt đầu được sử dụng trong điều trị bệnh phong. Dapsone được phát triển độc lập bởi hai nhà khoa học là Ernest Fourneau tại Pháp và Gladwin Buttle tại Vương quốc Anh. Ngoài sử dụng trong điều trị bệnh phong, Dapsone cũng được phối hợp với Chlorproguanil trong điều trị bệnh sốt rét.
Thalidomide vốn là một thuốc an thần, trước đây từng được sử dụng cho điều trị các triệu chứng ốm nghén trong thời kỳ mang thai. Tuy nhiên, sau này người ta phát hiện ra Thalidomide gây ra các dị tật thai nhi (“Thảm họa Thalidomide”), vậy nên nó đã bị hạn chế sử dụng. Thuốc có tác dụng khá tốt trên bệnh phong, đặc biệt là thể phong củ.
Cấu trúc hóa học
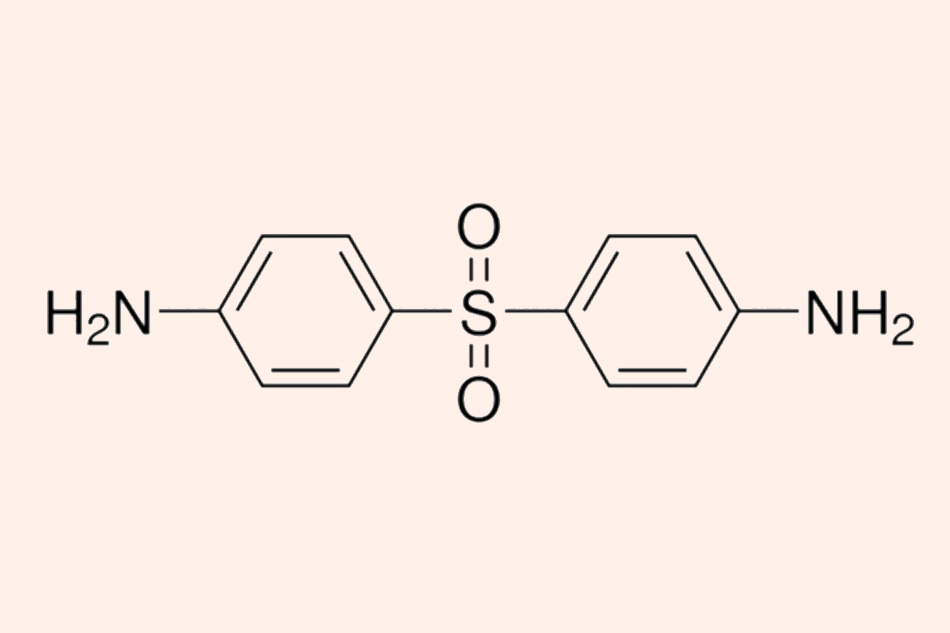
Đây là cấu trúc hóa học của Dapsone.
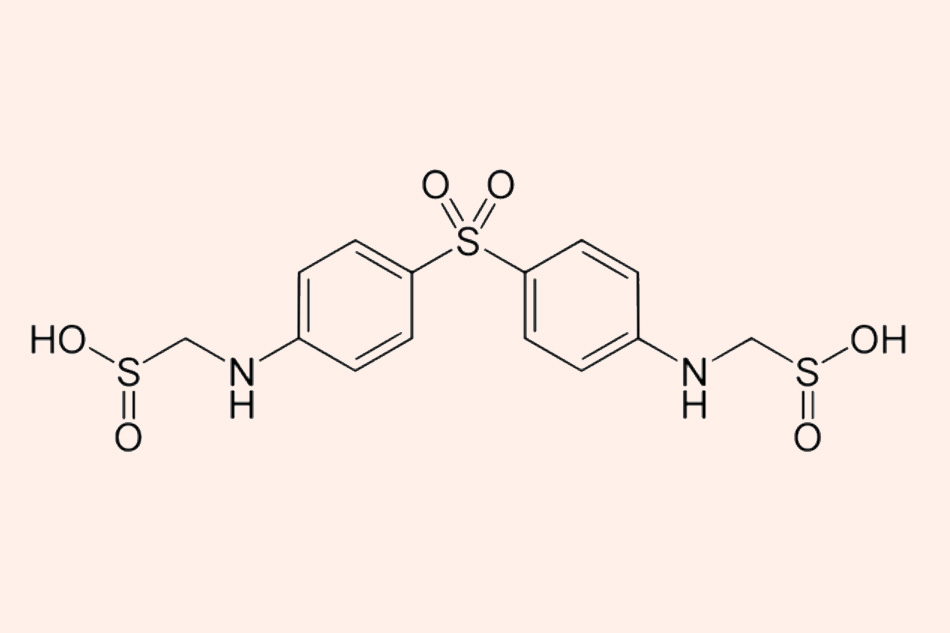
Đây là cấu trúc hóa học của Sulfoxone tương tự với Dapsone.
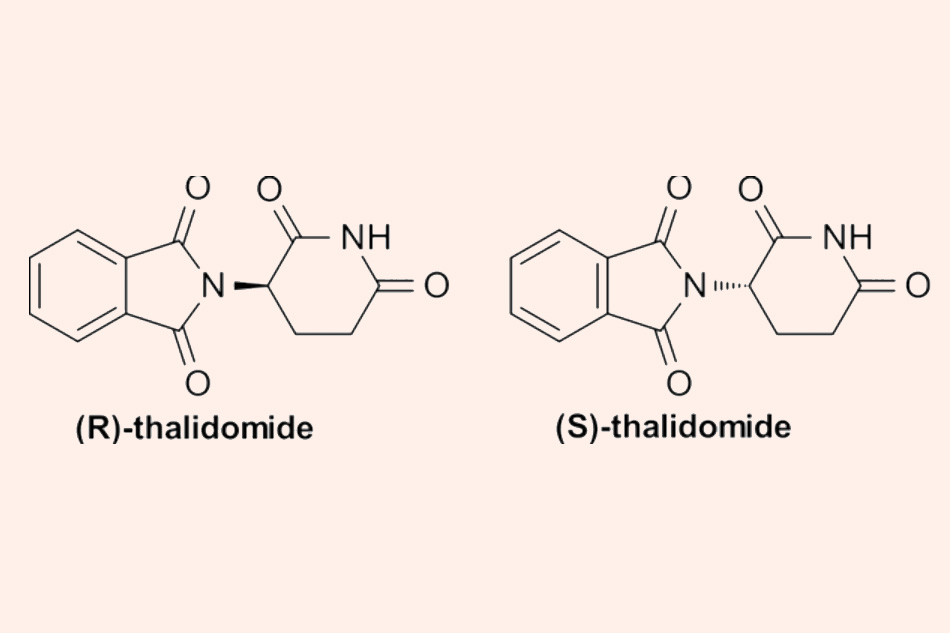
Đây là cấu trúc hóa học của Thalidomide. Thuốc có hai đồng phân quang học R và S.
Dược lý học
Dược lực học
Cơ chế tác dụng
Dapsone và Sulfoxone: Cả hai thuốc này đều có cơ chế tác dụng giống nhau và tương tự như các kháng sinh nhóm Sulfamide (Sulfonamide), đó là ức chế tổng hợp folate của vi khuẩn bằng cách ức chế cạnh tranh enzyme dihydropteroate synthase do cấu trúc của nó khá tương đồng với PABA (para-aminobenzoic acid).
Ngoài tác dụng kháng khuẩn, các thuốc này còn có tác dụng chống viêm tương tự như các thuốc kháng viêm không steroid (NSAIDs). Tuy nhiên, tác dụng chống viêm sẽ không được trình bày tại đây.
Thalidomide: Không rõ cơ chế tác dụng của thuốc trên trực khuẩn phong. Tuy nhiên, người ta đã biết Thalidomide có tác dụng điều biến nồng độ của cytokine tiền viêm TNF-α, điều này đem lại lợi ích trong điều trị phong củ. Ngoài ra, thuốc cũng có thể điều biến sự sản xuất interleukin (IL)-10 và IL-12.
Cơ chế đề kháng
- Dapsone và Sulfoxone: Đột biến gene folP1 mã hóa cho enzyme dihydropteroate synthase có liên quan đến đề kháng Dapsone và cả Sulfoxone. Hai đột biến sai lệch trên folP1 đã được xác định, đó là đột biến trên codon 53, thay thế threonine thành isoleucine và đột biến trên codon 55, thay thế proline thành arginine.
- Thalidomide: Không rõ.
Dược động học
Dapsone
- Hấp thu: Hấp thu gần như hoàn toàn qua đường tiêu hóa với sinh khả dụng trên 86%. Tmax thay đổi từ 2-8 giờ.
- Phân bố: Dapsone được phân bố tốt đến hầu hết các cơ quan. Các cơ quan lưu giữ nhiều Dapsone là da, cơ, thận và gan. Dapsone có liên kết với protein huyết tương dao động từ 50-90%. Chất chuyển hóa Monoacteyldapsone của thuốc liên kết với protein huyết tương gần như 100%. Dapsone có khả năng đi qua được hàng rào máu não, hàng rào nhau thai và qua được sữa mẹ.
- Chuyển hóa: Thuốc được chuyển hóa tại gan, đồng thời cũng được hoạt hóa nhờ bạch cầu đa nhân trung tính và bạch cầu đơn nhân. Tại gan, thuốc được acetyl hóa nhờ enzyme N-acetyltransferase thành Monoacteyldapsone, và được hydroxyl hóa nhờ các enzyme của CYP450 tại gan, tạo thành Dapsone hydroxylamine. Con đường chuyển hóa thành Dapsone hydroxylamine cũng xảy ra tại vùng da tổn thương do viêm da. Sau các phản ứng trên, tại gan thuốc tiếp tục được liên hợp với glucuronic acid nhờ enzyme uridine diphosphate (UDP)-glucuronosyltransferase.
- Thải trừ: Dapsone được bài xuất qua nước tiểu 20% dưới dạng không đổi và 70-85% dưới dạng chất chuyển hóa liên hợp glucuronic.
Thalidomide
Chỉ có dữ liệu dược động học trên người tình nguyện khỏe mạnh.
- Hấp thu: Thuốc được hấp thu chậm, Tmax khoảng 2-5 giờ.
- Phân bố: Liên kết với protein huyết tương của đồng phân R là 55% và đồng phân S là 66%.
- Thải trừ: t1/2 khoảng 5.5-7.3 giờ. Thuốc được bài xuất qua nước tiểu dưới dạng chủ yếu là chất chuyển hóa thủy phân.
Tác dụng – Chỉ định
Các thuốc Dapsone, Clofazimine và Rifampicin được chỉ định đầu tay cho bệnh phong. Các thuốc Ethionamide, Prothionamide, Sulfoxone và Thalidomide chỉ được chỉ định khi các thuốc đầu tay không hiệu quả.
Cách dùng – Liều dùng
- Dapsone: 100 mg/ngày.
- Clofazimine: 50 mg/ngày với phong nhạy cảm Dapsone và 100 mg/ngày với phong kháng Dapsone.
- Rifampicin: 600 mg/ngày.
- Sulfoxone: Thuốc đã không còn được lưu hành tại châu Âu, Hoa Kỳ hoặc Việt Nam.
- Thalidomide: 100-300 mg/ngày. Nên uống trước khi đi ngủ, ít nhất 1 giờ sau bữa tối. Liều cao hơn, lên đến 400 mg/ngày cũng có thể được cân nhắc tùy theo tình trạng bệnh. Khi các triệu chứng bệnh cải thiện, có thể giảm liều 50 mg mỗi 2-4 tuần.
Tác dụng không mong muốn
- Dapsone: Thuốc gây ra thiếu máu tan máu ở những bệnh nhân thiếu hụt enzyme G6PD (glucose-6 phosphate dehydrogenase). Các tác dụng không mong muốn khác có thể gặp phải bao gồm bệnh lý thần kinh ngoại biên, yếu cơ, rối loạn tiêu hóa (buồn nôn, nôn, viêm tụy…), mờ mắt, chóng mặt, ù tai, mất ngủ, hội chứng thận hư, giảm albumin máu, nhạy cảm ánh sáng…
- Thalidomide: Thuốc có nhiều tác dụng không mong muốn, bao gồm gây quái thai, huyết khối động và tĩnh mạch, buồn ngủ, chóng mặt, bệnh lý thần kinh ngoại biên, hạ huyết áp thế đứng, giảm bạch cầu trung tính hoặc giảm tiểu cầu, nhịp chậm, các phản ứng dị ứng trên da nghiêm trọng hoặc hội chứng ly giải khối u.
Chống chỉ định
Quá mẫn cảm với bất cứ thành phần nào của thuốc.
- Dapsone: Quá mẫn cảm với các dẫn xuất của Dapsone (ví dụ: Sulfoxone).
- Thalidomide: Phụ nữ có thai.
Tương tác thuốc
Dapsone:
- Phối hợp với các thuốc làm tăng pH dạ dày (thuốc kháng acid dịch vị, thuốc ức chế bơm proton hoặc thuốc kháng histamine H2): Hấp thu của Dapsone giảm do tăng pH dịch vị. Cần đảm bảo thời gian giữa các lần sử dụng các thuốc này cách xa nhau.
- Phối hợp với thuốc cảm ứng hoặc ức chế CYP3A4 mạnh: Nồng độ dapsone trong máu sẽ bị giảm hoặc tăng tương ứng, gây ra giảm tác dụng điều trị hoặc tăng nguy cơ gặp phải độc tính của thuốc, tương ứng.
- Phối hợp với vaccin thương hàn sống hoặc BCG sống: Không sử dụng phối hợp này do tác dụng của vaccin sẽ giảm đi.
- Phối hợp với các thuốc kháng acid folic khác (ví dụ: Methotrexate): Tăng nguy cơ gặp các tác dụng không mong muốn trên hệ tạo máu.
Thalidomide:
- Phối hợp với các thuốc có tác dụng an thần (thuốc kháng histamine H1, thuốc chống loạn thần, thuốc chống trầm cảm…): Tăng tác dụng an thần. Nên tránh phối hợp này.
- Phối hợp với các thuốc có tác dụng hạ nhịp tim (thuốc chẹn β-adrenergic, thuốc chẹn kênh calcium nhóm non-dihydropyridine…): Tăng tác dụng làm chậm nhịp tim. Thận trọng với phối hợp này.
- Phối hợp với thuốc tránh thai hormon chứa estrogen: Tăng nguy cơ huyết khối. Nên tránh phối hợp này.
Lưu ý và thận trọng khi sử dụng thuốc
Dapsone: Thận trọng khi điều trị cho các bệnh nhân có tiền sử quá mẫn với các Sulfamide. Đã có các trường hợp tử vong liên quan đến độc tính trên hệ tạo máu của Dapsone như mất bạch cầu hạt hoặc thiếu máu bất sản, giám sát cẩn thận.
Thalidomide:
- Thuốc có độc tính cao với phôi thai. Không sử dụng thuốc cho phụ nữ có thai hoặc có ý định có thai. Phụ nữ đang trong độ tuổi sinh sản sử dụng Thalidomide cũng cần áp dụng các biện pháp tránh thai an toàn.
- Xem xét điều trị dự phòng huyết khối dựa trên các yếu tố nguy cơ của bệnh nhân.
- Tổn thương thần kinh do Thalidomide gây ra có thể là vĩnh viễn. Theo dõi thận trọng.
- Nếu lượng bạch cầu trung tính dưới 750 tế bào/mm3, không nên dùng Thalidomide. Bệnh nhân cũng cần được theo dõi công thức máu cho giảm tiểu cầu có thể xảy ra.
- Thận trọng khi bệnh nhân phải thực hiện các công việc nguy hiểm, đòi hỏi tập trung cao như làm việc trên cao, lái xe, vận hành máy móc.
- Thuốc làm tăng tải lượng HIV trong máu so với giả dược có ý nghĩa thống kê trong một thử nghiệm lâm sàng có đối chứng ngẫu nhiên.
- Các phản ứng dị ứng trên da nghiêm trọng như hội chứng Stevens-Johnson hay tiêu thượng bì nhiễm độc có thể xảy ra.
- Co giật, kể cả co giật mức độ nặng, đã được báo cáo hậu mãi.
Sử dụng thuốc trên đối tượng đặc biệt
Dapsone:
- Phân loại an toàn trong thai kỳ: C. Chỉ sử dụng thuốc trên phụ nữ có thai khi thực sự cần thiết.
- Với phụ nữ đang cho con bú, không khuyến cáo sử dụng thuốc do thuốc có thể vào được sữa mẹ. Nếu bắt buộc phải dùng thuốc cho người mẹ, trẻ nên được cho dùng sữa ngoài.
Thalidomide:
- Phụ nữ có thai: Chống chỉ định.
- Phụ nữ đang cho con bú: Không có thông tin về sự hiện diện của Thalidomide trong sữa mẹ. Không khuyến cáo sử dụng thuốc khi đang cho con bú, cân nhắc cho trẻ dùng sữa ngoài nếu bắt buộc phải sử dụng thuốc cho người mẹ.
Một số nghiên cứu và thử nghiệm lâm sàng
Thử nghiệm lâm sàng so sánh Ofloxacin đơn độc hoặc kết hợp với Dapsone và Clofazimine trong điều trị bệnh phong thể u cho thấy: Sự cải thiện lâm sàng, hoạt tính kháng khuẩn và độc tính trên gan là tương tự nhau ở cả ba nhóm dùng 56 ngày Ofloxacin 400 mg/ngày, Ofloxacin 800 mg/ngày hoặc Ofloxacin 400 mg, Dapsone 100 mg và Clofazimine 50 mg hàng ngày cộng với Clofazimine 300 mg một lần trong 28 ngày. Ofloxacin có hoạt tính chống trực khuẩn phong tốt và có thể trở thành một thành phần quan trọng trong phác đồ đa hóa trị mới dành cho bệnh phong. Liều tối ưu của Ofloxacin là 400 mg/ngày, kết hợp với Dapsone và Clofazimine không làm tăng tác dụng của nó.
Tài liệu tham khảo
Sotgiu G, Centis R, D’ambrosio L, Migliori GB. Tuberculosis treatment and drug regimens. Cold Spring Harb Perspect Med. 2015;5(5):a017822. Published 2015 Jan 8. doi: 10.1101/cshperspect.a017822.
Available at https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4448591/
Timmins GS, Deretic V. Mechanisms of action of isoniazid. Mol Microbiol. 2006 Dec;62(5):1220-7. doi: 10.1111/j.1365-2958.2006.05467.x.
Available at https://onlinelibrary.wiley.com/doi/full/10.1111/j.1365-2958.2006.05467.x
Wehrli W. Rifampin: mechanisms of action and resistance. Rev Infect Dis. 1983 Jul-Aug;5 Suppl 3:S407-11. doi: 10.1093/clinids/5.supplement_3.s407.
Available at https://academic.oup.com/cid/article-abstract/5/Supplement_3/S407/275556?redirectedFrom=PDF
Zhang Y, Shi W, Zhang W, Mitchison D. Mechanisms of Pyrazinamide Action and Resistance. Microbiol Spectr. 2013;2(4):1-12. doi: 10.1128/microbiolspec.
Available at https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4268777/
Lin Y, Li Y, Zhu N, et al. The antituberculosis antibiotic capreomycin inhibits protein synthesis by disrupting interaction between ribosomal proteins L12 and L10. Antimicrob Agents Chemother. 2014;58(4):2038-2044. doi: 10.1128/AAC.02394-13.
Available at https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4023752/
Maus CE, Plikaytis BB, Shinnick TM. Mutation of tlyA confers capreomycin resistance in Mycobacterium tuberculosis. Antimicrob Agents Chemother. 2005;49(2):571-7. doi: 10.1128/AAC.49.2.571-577.2005.
Available at https://www.ncbi.nlm.nih.gov/pmc/articles/PMC547314/
Kambli P, Ajbani K, Nikam C, et al. Correlating rrs and eis promoter mutations in clinical isolates of Mycobacterium tuberculosis with phenotypic susceptibility levels to the second-line injectables [published correction appears in Int J Mycobacteriol. 2016 Sep;5(3):370-372]. Int J Mycobacteriol. 2016;5(1):1-6. doi: 10.1016/j.ijmyco.2015.09.001.
Available at https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4863938/
Wang F, Langley R, Gulten G, et al. Mechanism of thioamide drug action against tuberculosis and leprosy. J Exp Med. 2007;204(1):73-78. doi: 10.1084/jem.20062100.
Available at https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2118422/
Zhang Y. The magic bullets and tuberculosis drug targets. Annu Rev Pharmacol Toxicol. 2005;45:529-64. doi: 10.1146/annurev.pharmtox.45.120403.100120.
Available at https://pubmed.ncbi.nlm.nih.gov/15822188/
Ramaswamy S, Musser JM. Molecular genetic basis of antimicrobial agent resistance in Mycobacterium tuberculosis: 1998 update. Tuber Lung Dis. 1998;79(1):3-29. doi: 10.1054/tuld.1998.0002.
Available at https://pubmed.ncbi.nlm.nih.gov/10645439/
Pandey B, Grover S, Kaur J, Grover A. Analysis of mutations leading to para-aminosalicylic acid resistance in Mycobacterium tuberculosis. Sci Rep. 2019 Sep 20;9(1):13617. doi: 10.1038/s41598-019-48940-5.
Available at https://www.nature.com/articles/s41598-019-48940-5
Benoit Lechartier, Stewart T. Cole. Mode of Action of Clofazimine and Combination Therapy with Benzothiazinones against Mycobacterium tuberculosis. Antimicrob Agents Chemother. 2015;59(8):4457-63. doi: 10.1128/AAC.00395-15.
Available at https://aac.asm.org/content/59/8/4457
Yew WW, Liang D, Chan DP, Shi W, Zhang Y. Molecular mechanisms of clofazimine resistance in Mycobacterium tuberculosis. J Antimicrob Chemother. 2017 Oct 1;72(10):2943-44. doi: 10.1093/jac/dkx227.
Available at https://academic.oup.com/jac/article/72/10/2943/3979532
Alahari A, Trivelli X, Guérardel Y, Dover LG, Besra GS, Sacchettini JC, Reynolds RC, Coxon GD, Kremer L. Thiacetazone, an antitubercular drug that inhibits cyclopropanation of cell wall mycolic acids in mycobacteria. PLoS One. 2007 Dec 19;2(12):e1343. doi: 10.1371/journal.pone.0001343.
Available at https://pubmed.ncbi.nlm.nih.gov/18094751/
Alahari A, Alibaud L, Trivelli X, Gupta R, Lamichhane G, et al. Mycolic acid methyltransferase, MmaA4, is necessary for thiacetazone susceptibility in Mycobacterium tuberculosis. Mol Microbiol. 2009;71:1263–77. doi: 10.1111/j.1365-2958.2009.06604.x.
Available at https://onlinelibrary.wiley.com/doi/full/10.1111/j.1365-2958.2009.06604.x
Belardinelli JM, Morbidoni HR. Mutations in the essential FAS II β-hydroxyacyl ACP dehydratase complex confer resistance to thiacetazone in Mycobacterium tuberculosis and Mycobacterium kansasii. Mol Microbiol. 2012 Nov;86(3):568-79. doi: 10.1111/mmi.12005.
Available at https://pubmed.ncbi.nlm.nih.gov/22994892/
Grzegorzewicz AE, Kordulakova J, Jones V, Born SE, Belardinelli JM, et al. A common mechanism of inhibition of the Mycobacterium tuberculosis mycolic acid biosynthetic pathway by isoxyl and thiacetazone. J Biol Chem. 2012;287:38434–41. doi: 10.1074/jbc.M112.400994.
Available at https://www.jbc.org/article/S0021-9258(20)62309-2/fulltext
Ordway D, Viveiros M, Leandro C, et al. Clinical concentrations of thioridazine kill intracellular multidrug-resistant Mycobacterium tuberculosis. Antimicrob Agents Chemother. 2003;47(3):917-922. doi: 10.1128/aac.47.3.917-922.2003.
Available at https://www.ncbi.nlm.nih.gov/pmc/articles/PMC149316/
Martins M, Dastidar SG, Fanning S, Kristiansen JE, Molnar J, Pagès JM, Schelz Z, Spengler G, Viveiros M, Amaral L. Potential role of non-antibiotics (helper compounds) in the treatment of multidrug-resistant Gram-negative infections: mechanisms for their direct and indirect activities. Int J Antimicrob Agents. 2008 Mar;31(3):198-208. doi: 10.1016/j.ijantimicag.2007.10.025.
Available at https://pubmed.ncbi.nlm.nih.gov/18180147/
Dutta NK, Mehra S, Kaushal D. A Mycobacterium tuberculosis sigma factor network responds to cell-envelope damage by the promising anti-mycobacterial thioridazine. PLoS One. 2010;5(4):e10069. Published 2010 Apr 8. doi: 10.1371/journal.pone.0010069.
Available at https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2851646/
Amaral L, Kristiansen JE, Viveiros M, Atouguia J. Activity of phenothiazines against antibiotic-resistant Mycobacterium tuberculosis: a review supporting further studies that may elucidate the potential use of thioridazine as anti-tuberculosis therapy. J Antimicrob Chemother. 2001 May;47(5):505-11. doi: 10.1093/jac/47.5.505.
Available at https://academic.oup.com/jac/article/47/5/505/858497
Cholo MC, Mothiba MT, Fourie B, Anderson R. Mechanisms of action and therapeutic efficacies of the lipophilic antimycobacterial agents clofazimine and bedaquiline. J Antimicrob Chemother. 2017 Feb;72(2):338-353. doi: 10.1093/jac/dkw426.
Available at https://academic.oup.com/jac/article/72/2/338/2290946
Sarathy JP, Gruber G, Dick T. Re-Understanding the Mechanisms of Action of the Anti-Mycobacterial Drug Bedaquiline. Antibiotics (Basel). 2019;8(4):261. Published 2019 Dec 11. doi: 10.3390/antibiotics8040261.
Available at https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6963887/
Lewis JM, Sloan DJ. The role of delamanid in the treatment of drug-resistant tuberculosis. Ther Clin Risk Manag. 2015;11:779-791. Published 2015 May 13. doi: 10.2147/TCRM.S71076.
Available at https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4437614/
Xavier AS, Lakshmanan M. Delamanid: A new armor in combating drug-resistant tuberculosis. J Pharmacol Pharmacother. 2014;5(3):222-224. doi: 10.4103/0976-500X.136121.
Available at https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4156838/
Zítková L, Tousek J. Pharmacokinetics of cycloserine and terizidone. A comparative study. Chemotherapy. 1974;20(1):18-28. doi: 10.1159/000221787.
Available at https://pubmed.ncbi.nlm.nih.gov/4845674/
Peloquin CA, Nitta AT, Berning SE, Iseman MD, James GT. Pharmacokinetic evaluation of thiacetazone. Pharmacotherapy. 1996 Sep-Oct;16(5):735-41.
Available at https://pubmed.ncbi.nlm.nih.gov/8888068/
Charan J, Reljic T, Kumar A. Bedaquiline versus placebo for management of multiple drug-resistant tuberculosis: A systematic review. Indian J Pharmacol. 2016;48(2):186-191. doi: 10.4103/0253-7613.178839.
Available at https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4825437/
Conradie F, Diacon AH, Ngubane N, et al. Treatment of Highly Drug-Resistant Pulmonary Tuberculosis. N Engl J Med. 2020;382(10):893-902. doi: 10.1056/NEJMoa1901814.
Available at https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6955640/
Wozel G, Blasum C. Dapsone in dermatology and beyond. Arch Dermatol Res. 2014;306(2):103-124. doi: 10.1007/s00403-013-1409-7.
Available at https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3927068/
Teo S, Resztak KE, Scheffler MA, Kook KA, Zeldis JB, Stirling DI, Thomas SD. Thalidomide in the treatment of leprosy. Microbes Infect. 2002 Sep;4(11):1193-202. doi: 10.1016/s1286-4579(02)01645-3.
Available at https://pubmed.ncbi.nlm.nih.gov/12361920/
Williams DL, Spring L, Harris E, Roche P, Gillis TP. Dihydropteroate synthase of Mycobacterium leprae and dapsone resistance [published correction appears in Antimicrob Agents Chemother 2001 Feb;45(2):647]. Antimicrob Agents Chemother. 2000;44(6):1530-1537. doi: 10.1128/aac.44.6.1530-1537.2000.
Available at https://www.ncbi.nlm.nih.gov/pmc/articles/PMC89908/
Ji B, Perani EG, Petinom C, N’Deli L, Grosset JH. Clinical trial of ofloxacin alone and in combination with dapsone plus clofazimine for treatment of lepromatous leprosy. Antimicrob Agents Chemother. 1994 Apr;38(4):662-7. doi: 10.1128/aac.38.4.662.
Available at https://pubmed.ncbi.nlm.nih.gov/8031029/
Xem thêm: Kháng sinh nhóm Pleuromutilin: Tác dụng, Chỉ định, Tác dụng phụ




{kind=link}