Giới thiệu chung về Kháng sinh nhóm Glycopeptide
Lịch sử ra đời

Glycopeptide là một nhóm kháng sinh mạnh và quan trọng trên lâm sàng. Chúng là những vũ khí quan trọng của con người trong cuộc chiến chống lại các vi khuẩn gram dương đa kháng thuốc. Hiện tại, nhóm kháng sinh này bao gồm năm thuốc (còn được sử dụng trên lâm sàng): Vancomycin và Teicoplanin là hai kháng sinh có nguồn gốc từ tự nhiên, Telavancin, Dalbavancin và Oritavancin là các kháng sinh bán tổng hợp.
Hiện tại, các vi khuẩn đa kháng thuốc đang là một mối lo ngại lớn trên toàn cầu, không chỉ ảnh hưởng tới các nước đang và kém phát triển, mà chúng cũng ảnh hưởng rất sâu sắc đến cả các nước phát triển như Anh hoặc Hoa Kỳ. Đi cùng với tình trạng kháng kháng sinh đáng báo động của các vi khuẩn thì sự phát triển các kháng sinh mới lại đang ngày càng chậm lại. Các vi khuẩn kháng thuốc nhiều nhất phần lớn tập trung về phía các vi khuẩn gram âm như Acinetobacter baumannii, Pseudomonas aeruginosa (trực khuẩn mủ xanh), Klebsiella pneumoniae, E.coli và các chủng Enterobacteriaceae kháng Carbapenem sinh ESBL (Extended-Spectrum β-lactamase: β-lactamase phổ mở rộng), ngoài ra cũng có một số vi khuẩn gram dương kháng thuốc cực mạnh, với điển hình là MRSA (Methicillin-Resistant Staphylococcus aureus: Tụ cầu vàng kháng Methicillin), các cầu khuẩn ruột Enterococcus (bao gồm hai loài kháng thuốc nổi bật nhất là E.faecium và E.faecalis) và Streptococcus pneumoniae (phế cầu).
Các liệu pháp kháng sinh hiện nay được áp dụng phổ biến cho các nhiễm trùng do vi khuẩn gram dương đa kháng gây ra bao gồm: Glycopeptide (với năm đại diện đã nói ở trên), Lipopeptide (Daptomycin), Oxazolidinone (Linezolid và Tedizolid), Cephalosporin thế hệ năm (Ceftaroline, Ceftolozane và Ceftobiprole) và Glycylcycline (Tigecycline). Bài viết này chỉ tập trung vào các Glycopeptide.
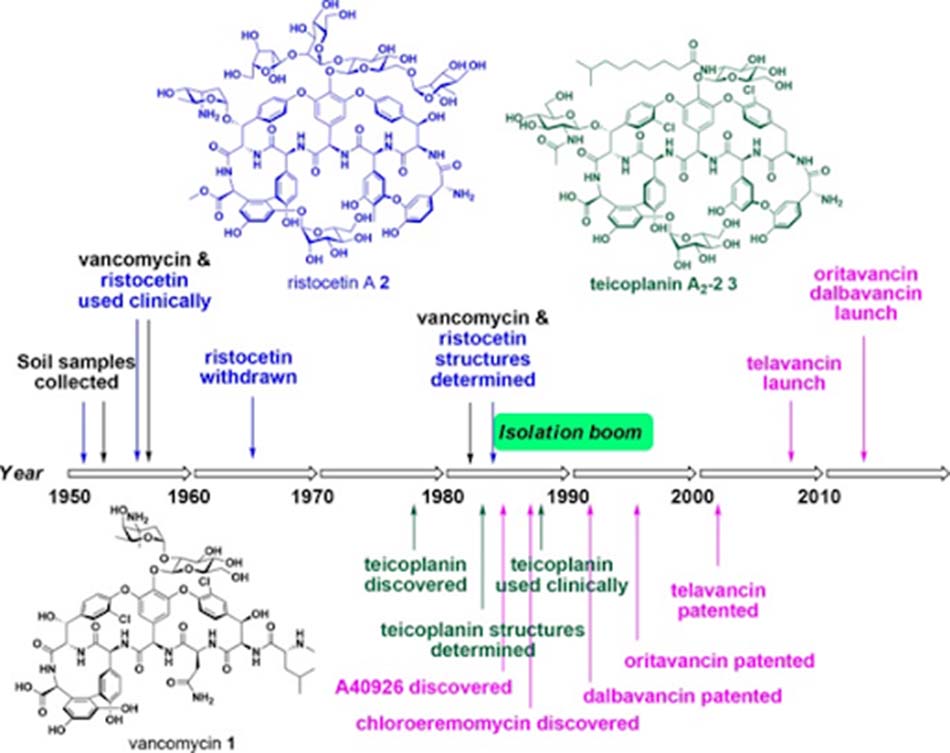
Hai loại kháng sinh nhóm Glycopeptide được phát hiện ra đầu tiên có nguồn gốc từ xạ khuẩn là Vancomycin và Ristocetin (giữa những năm 1950). Chúng được phát hiện lần lượt bởi Eli Lilly và Abbott Laboratories từ các loài tương ứng là Amycolatopsis orientalis và A.lurida. Cả hai loại kháng sinh này đều đã được phê duyệt để sử dụng trên lâm sàng, nhưng sau đó không lâu, Ristocetin đã phải bị thu hồi do nó gây giảm tiểu cầu. Vancomycin được sử dụng lần đầu trên người năm 1955, chính thức được phê duyệt năm 1958, nhưng phải đến năm 1982, cấu trúc hóa học của nó mới được xác định. Vào những năm 1960 và 1970, thời kỳ vàng son của kháng sinh, cùng thời với Vancomycin, còn có nhiều nhóm kháng sinh mới khác cũng được phát hiện và có nhiều ưu điểm hơn Vancomycin (nhiều thuốc có thể sử dụng theo đường uống, có dược động học thuận lợi, phổ tác dụng tốt và ít tác dụng không mong muốn). Chính vì vậy, việc sử dụng Vancomycin là không phổ biến cho đến những năm 1980, khi tỷ lệ các vi khuẩn đề kháng với nhiều β-lactam tăng lên đáng kể, cùng với đó là sự gia tăng tỷ lệ MRSA trong bệnh viện. Vào thời điểm đó, do Vancomycin chưa được sử dụng nhiều nên tỷ lệ kháng thuốc rất thấp, do đó nó đã được được sử dụng phổ biến trở lại trên lâm sàng, cho đến khi tình trạng kháng Vancomycin bắt đầu tăng dần.
Ngoài Vancomycin và Ristocetin, nhóm Glycopeptide còn một kháng sinh có nguồn gốc từ tự nhiên khác, đó là Teicoplanin. Teicoplanin là một phức hợp Lipoglycopeptide type Ristocetin được báo cáo lần đầu năm 1978, được phân lập từ loài Actinoplanes teichomyceticus. Thuốc này đã được phê duyệt ở châu Âu năm 1988 và Nhật bản năm 1998, nhưng chưa từng được phê duyệt tại Hoa Kỳ.
Giai đoạn 1982-1996 là giai đoạn mà rất nhiều kháng sinh Glycopeptide mới được phát hiện, nhưng đã không có kháng sinh nào có thể được đưa vào sử dụng trên lâm sàng. Tuy vậy, sau giai đoạn này, số lượng các phân tử kháng sinh Glycopeptide tự nhiên mới được phát hiện đã giảm đáng kể, chủ yếu là do các chủng xạ khuẩn dùng để sản xuất Glycopeptide không phổ biến. Từ thời điểm này trở đi, các kháng sinh Glycopeptide được nghiên cứu và phát triển chủ yếu là các dẫn chất bán tổng hợp.
Telavancin là một kháng sinh nhóm Glycopeptide (thực chất là Lipoglycopeptide) thế hệ hai, được ra mắt bởi Theravance năm 2009 dưới tên biệt dược Vibativ. Telavancin là dẫn chất của Vancomycin, với đuôi decylaminoethyl thân dầu được gắn trên đường vancosamine và nhóm (phosphonomethyl)aminomethyl thân nước ở vị trí 4’ trên vòng thơm của amino acid 7. Sự cải tiến về mặt công thức này cũng dẫn đến sự cải thiện đáng kể hoạt phổ trên vi khuẩn gram dương, cũng như cải thiện các đặc tính dược động học.
Dalbavancin là một Glycopeptide thế hệ hai type Teicoplanin, được phát triển bởi Durata Therapeutics/Allergan dưới tên thương mại Dalvance. Quá trình phát triển của thuốc này kéo dài tới hơn 15 năm, cuối cùng nó đã được phê duyệt năm 2014. Nó là một dẫn chất của A40926, một Glycopeptide tự nhiên, được tạo ra bằng cách amide hóa nhóm carboxy của amino acid 7 bằng 3-(dimethylamino)-1-propylamine.
Cuối cùng, Oritavancin là một Lipoglycopeptide thế hệ hai, do Eli Lilly phát triển và The Medicines Company tiếp thị (đã được bán cho Melinta Therapeutics tháng 11/2017) dưới tên thương mại Orbactiv. Nó là dẫn xuất của Chloroeremomycin, một Glycopeptide tự nhiên. Nó được tạo ra bằng cách gắn nhóm thế N-alkyl-p-chlorophenylbenzyl trên epi-vancosamine của đường đôi (được gắn vào vòng 4 amino acid). Chloroeremomycin được Lilly phát hiện năm 1988, nó khác biệt với Vancomycin ở nhóm đường vancosamine, trong khi Chloroeremomycin chứa hai tiểu đơn vị L-4-epi-vancosamine trên vòng 4 và 6 amino acid, thì Vancomycin lại chỉ chứa một tiểu đơn vị L-vancosamine ở vị trí 4.
Tính đến thời điểm hiện tại (năm 2020), tại thị trường Việt Nam, mới chỉ có hai loại Glycopeptide được sử dụng trên lâm sàng là Vancomycin và Teicoplanin.
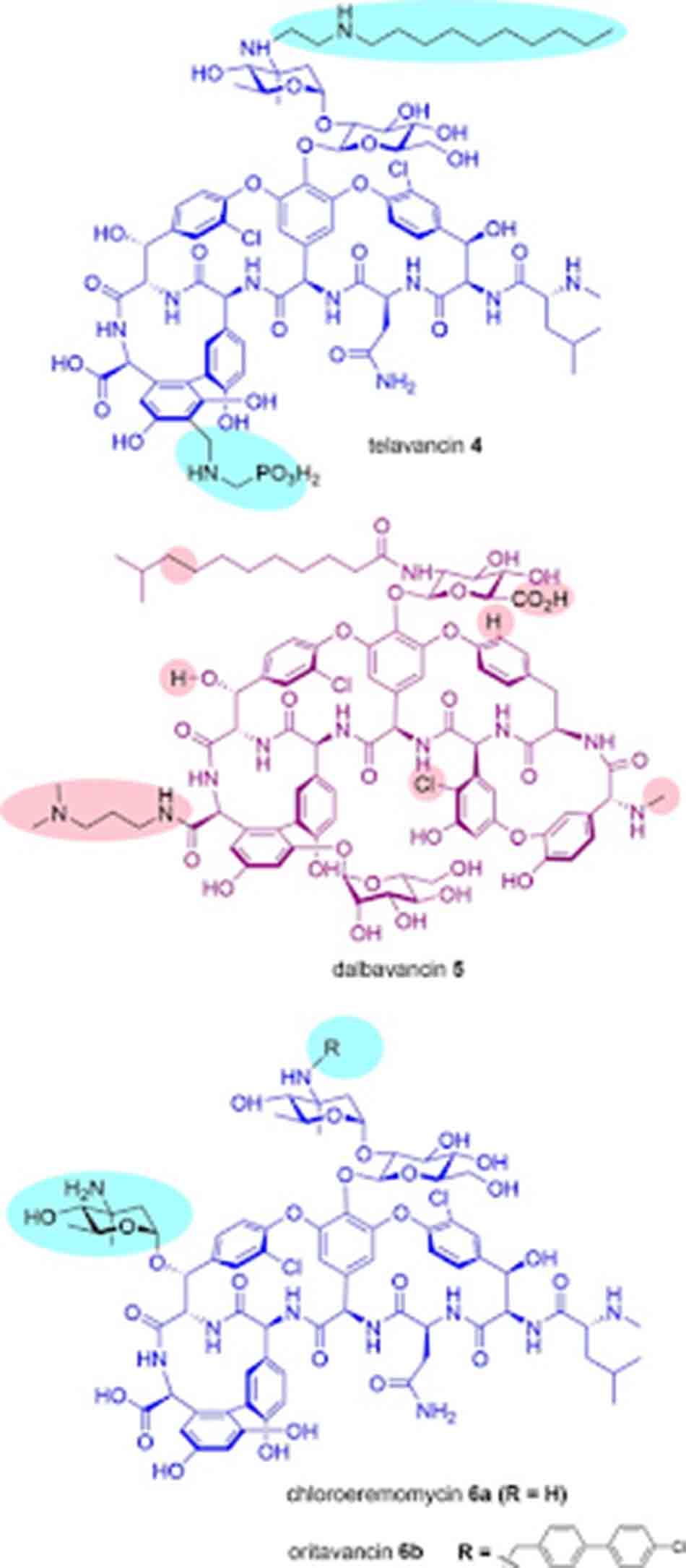
Cấu trúc hóa học
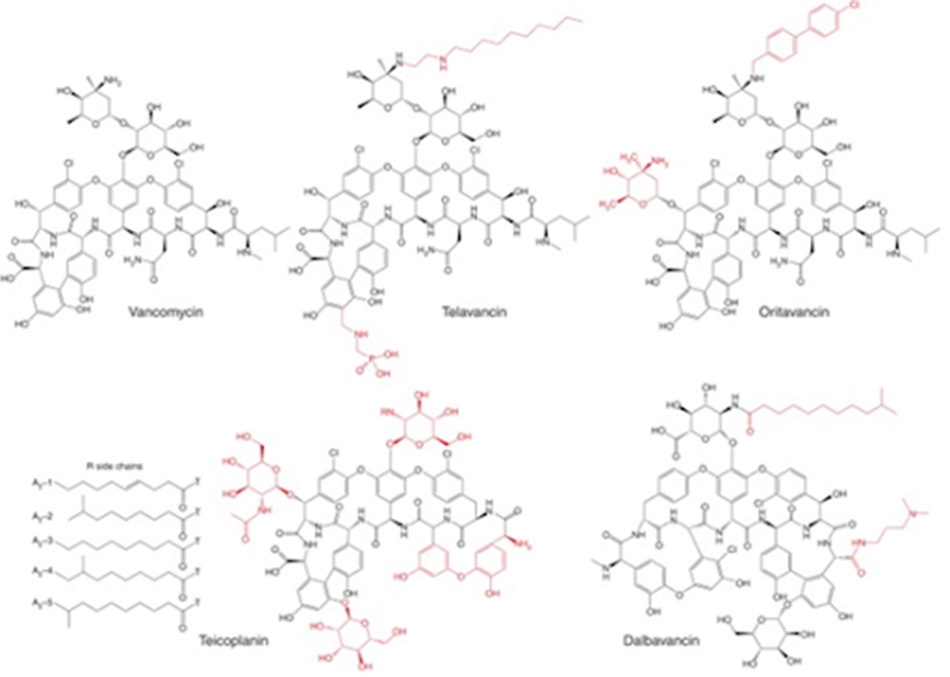
Dược lực học
Cơ chế tác dụng
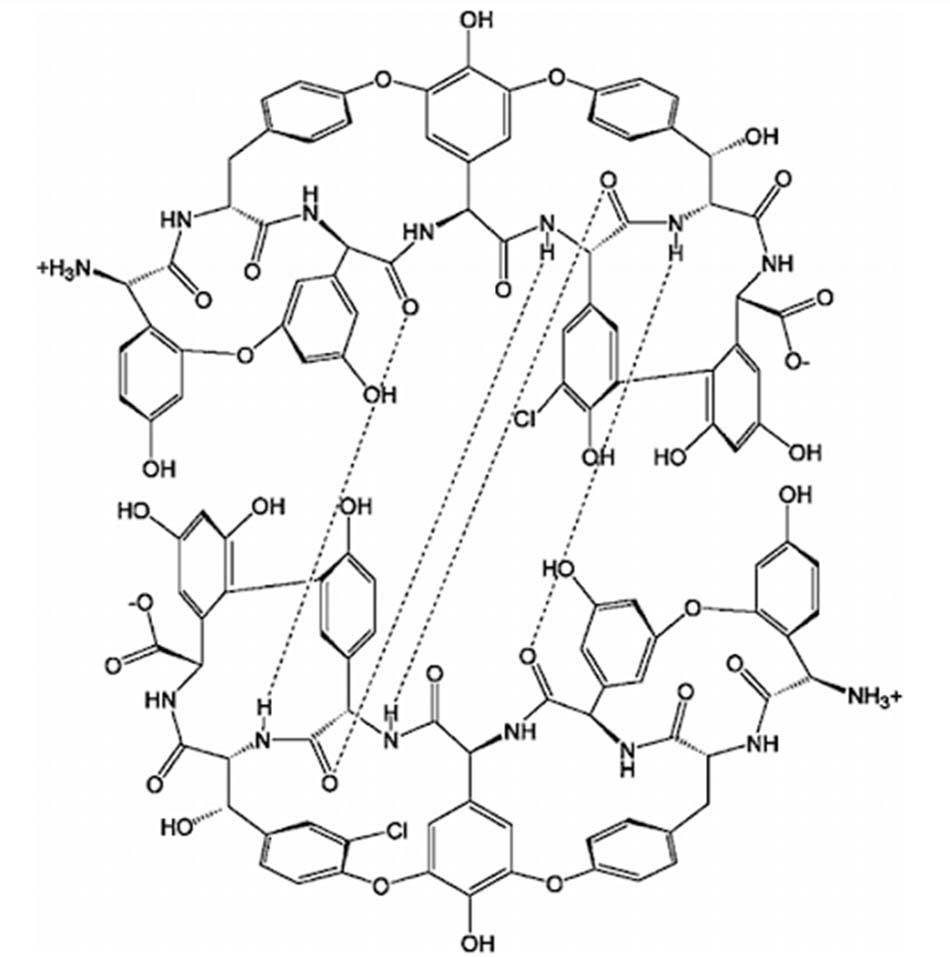
Các kháng sinh Glycopeptide đều có chung một cơ chế tác dụng giống nhau, đó là ức chế tổng hợp thành tế bào vi khuẩn. Cụ thể, các Glycopeptide ức chế tổng hợp lớp peptidoglycan, một thành phần quan trọng trong thành tế bào vi khuẩn. Peptidoglycan là một polymer có cấu trúc không gian ba chiều phức tạp, có độ bền vững cao, có tác dụng duy trì hình dạng vi khuẩn, bảo vệ vi khuẩn trước những tác nhân tấn công từ môi trường, đồng thời bảo vệ vi khuẩn khỏi bị vỡ bởi áp lực thẩm thấu nội bào rất cao. Không giống như các β-lactam ức chế tổng hợp lớp peptidoglycan của vi khuẩn thông qua ức chế transpeptidase, enzyme tham gia vào bước cuối cùng trong tổng hợp peptidoglycan, các Glycopeptide ức chế tổng hợp lớp peptidoglycan ở giai đoạn sớm hơn, cụ thể chúng liên kết với lipid II, thông qua các liên kết hydro với đầu D-Ala-D-Ala. Sự liên kết này làm ngăn cản quá trình transpeptidase hóa, từ đó cản trở quá trình hình thành các liên kết chéo trong lớp peptidoglycan.
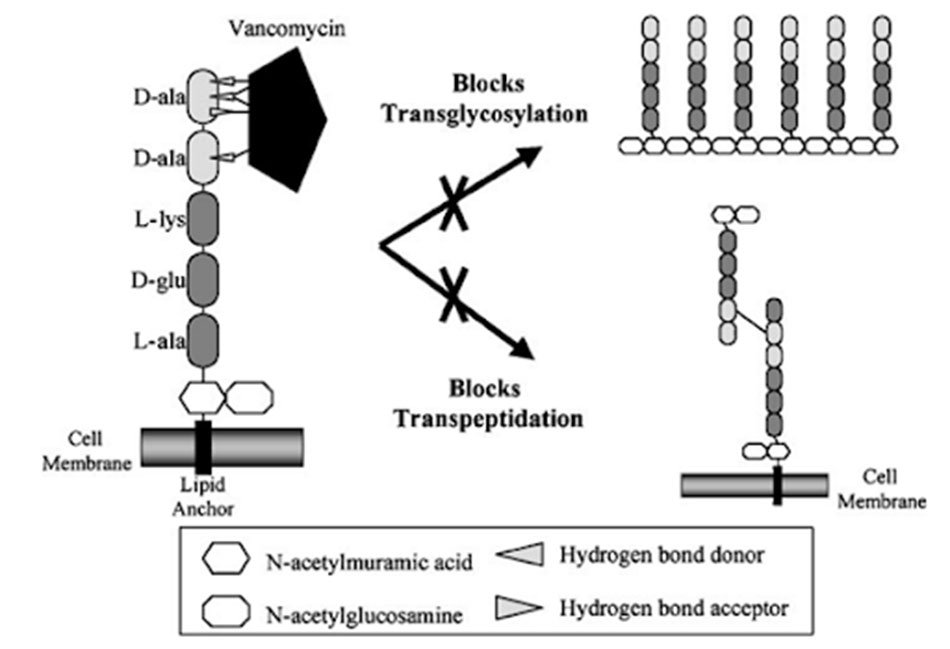
Mô tả: Kháng sinh gắn vào đầu D-Ala-D-Ala của cầu nối pentapeptide thông qua năm liên kết hydro, ức chế sự hình thành liên kết chéo của peptidoglycan cũng như ức chế quá trình transglycosyl hóa.
Cơ chế vừa nói ở trên là cơ chế tác dụng chung. Ngoài cơ chế này ra, một số phân tử kháng sinh có thêm một hoặc một vài cơ chế riêng biệt đặc trưng khác.
Vancomycin ức chế lipid II theo cách như đã nói ở trên, nhưng còn một điều đặc biệt là Vancomycin có khả năng tự dimer hóa, từ đó tăng cường liên kết với lipid II. Hoạt tính kháng khuẩn in vitro của Vancomycin dimer vượt trội hơn so với Vancomycin. Vancomycin cũng chống lại quá trình tái cấu trúc lớp peptidoglycan, còn gọi là quá trình “autolysis”.
Teicoplanin cũng có khả năng tự dimer hóa như Vancomycin, đồng thời nhờ có nhóm kỵ nước có khả năng tương tác với lớp lipid kép màng tế bào, phân tử Teicoplanin được “neo” gần vị trí của lipid II, tạo ra tương tác thuận lợi cho tác dụng kháng khuẩn.
Cơ chế hoạt động bổ sung của Dalbavancin được cho là tương tự Teicoplanin.
Oritavancin nhờ có chuỗi bên kỵ nước 4′-chlorobiphenylmethyl trên đường disaccharide mà có khả năng tương tác trực tiếp với màng tế bào vi khuẩn, làm cho quá trình tương tác giữa phân tử kháng sinh với lipid II ổn định hơn. Oritavancin cũng có khả năng hình thành dạng dimer tương tự như Vancomycin, làm cho liên kết với đầu D-Ala-D-Lac của vi khuẩn đề kháng Vancomycin được tăng cường (Điều này lý giải cho việc Oritavancin có tác dụng trên một số chủng vi khuẩn đã đề kháng Vancomycin). Ngoài ra, Oritavancin cũng đã được báo cáo là ức chế sự transpeptidase hóa. Nguyên nhân của cơ chế này có thể là do nhóm thế 4′-chlorobiphenylmethyl có khả năng gắn với cầu pentaglycyl. Nhóm thế này cũng được cho là nguyên nhân gây chết tế bào vi khuẩn do nó gây khử cực màng cũng như tăng tính thấm màng. Một điều thú vị là kháng sinh này cũng cho thấy có khả năng chống lại dạng biofilm (màng sinh học) của tụ cầu vàng in vitro. Cuối cùng, người ta còn tìm thấy Oritavancin có khả năng ức chế tổng hợp RNA, nhưng cơ chế này còn nhiều nghi vấn.
Telavancin không giống Vancomycin ở chỗ nó không ức chế quá trình “autolysis”. Nhưng giống như Oritavancin, Telavancin có khả năng gây khử cực màng tế bào nhanh và phụ thuộc nồng độ, làm tăng tính thấm màng tế bào cũng như làm rò rỉ ion kali và ATP – năng lượng của tế bào. Trong tác dụng ức chế quá trình transglycosyl hóa và tổng hợp peptidoglycan, Telavancin mạnh hơn Vancomycin 10 lần.
Một cơ chế khác của các lipoglycopeptide cũng được đề xuất là phần liposaccharide của lipoglycopeptide tương tác trực tiếp và ức chế enzyme transglycosylase (Enzyme trung gian cho quá trình polymer hóa các tiền chất tạo thành peptidoglycan chưa trưởng thành [chưa có liên kết chéo]). Tuy nhiên, có vẻ như cơ chế này không có ở Telavancin.
Phổ tác dụng
Phổ tác dụng của các kháng sinh Glycopeptide chỉ tập trung trên các vi khuẩn gram dương, đặc biệt là các loài Staphylococcus spp., Streptococcus spp. và Enterococcus spp. Trên các chủng tụ cầu Staphylococcus spp., chúng tác dụng tốt trên cả các chủng coagulase âm tính hoặc đã đề kháng với Methicillin, trong đó đặc biệt nổi tiếng là MRSA, một tác nhân gây nhiễm trùng bệnh viện phổ biến và nguy hiểm. Trên các chủng liên cầu Streptococcus spp., chúng tác dụng tốt trên cả các chủng đã đề kháng với Penicillin, phổ biến nhất là phế cầu S.pneumoniae. Trên cầu khuẩn ruột Enterococcus spp., chúng có tác dụng tốt với hai loại vi khuẩn đề kháng các kháng sinh nhóm khác mạnh là E.faecium và E.faecalis.
Ngoài ra, chúng cũng có tác dụng tốt trên các chủng vi khuẩn kỵ khí Clostridium spp., đặc biệt là vi khuẩn gây viêm đại tràng giả mạc C.difficile. Tác dụng của các kháng sinh nhóm này cũng tốt trên Listeria monocytogenes và Actinomyces.
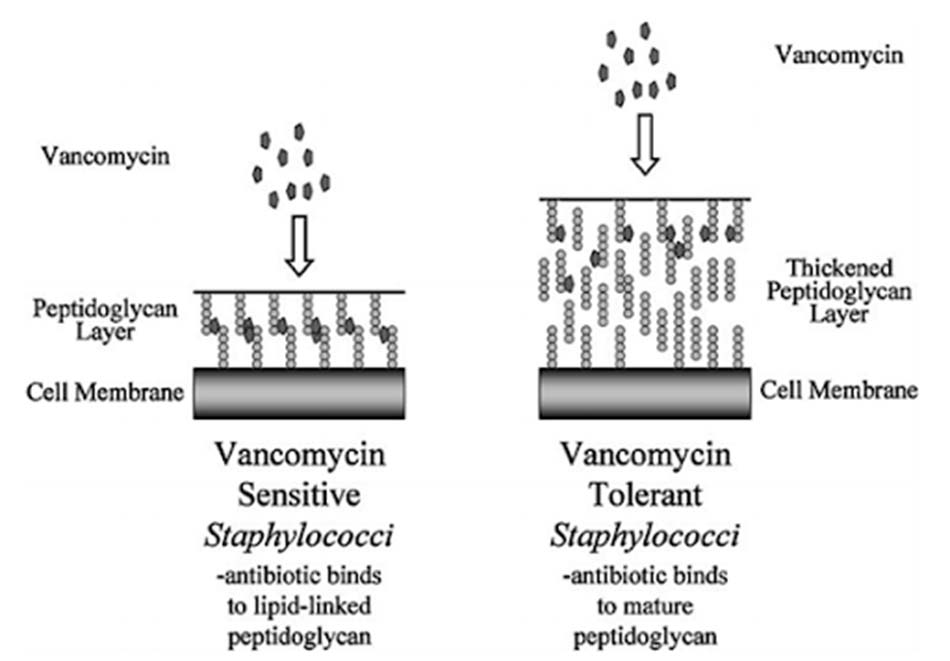
Hiện nay với việc sử dụng kháng sinh còn chưa hợp lý và tần suất ngày càng tăng cao, tình trạng đề kháng kháng sinh đang dần trở nên đáng báo động. Ngày càng nhiều chủng MRSA hoặc Enterococcus được phân lập cho thấy khả năng đề kháng Vancomycin, các chủng này được gọi là VRSA (Vancomycin-Resistant Staphylococcus aureus: Tụ cầu vàng kháng Vancomycin) và VRE (Vancomycin-Resistant Enterococcus: Enterococcus kháng Vancomycin). Teicoplanin và Dalbavancin cho thấy có khả năng chống lại Enterococcus VanB. Telavancin và Oritavancin còn cho thấy hoạt tính chống lại Enterococcus VanA, nhưng ý nghĩa lâm sàng của nó là không chắc chắn.
Lý do các kháng sinh nhóm Glycopeptide không có phổ tác dụng trên vi khuẩn gram âm là vì chúng không thể qua được lớp màng ngoài của vi khuẩn gram âm.
Cơ chế đề kháng
Trường hợp đầu tiên đầu tiên được báo cáo đề kháng với Vancomycin là vào năm 1987, tức khoảng 30 năm sau khi Vancomycin được sử dụng trên lâm sàng. Nguyên nhân chủ yếu là do tình trạng sử dụng Vancomycin trước đó bị hạn chế. Từ thời điểm đó cho đến nay, tình trạng đề kháng kháng sinh nhóm này đang tăng đều, đặc biệt là trên các chủng tụ cầu và cầu khuẩn ruột.
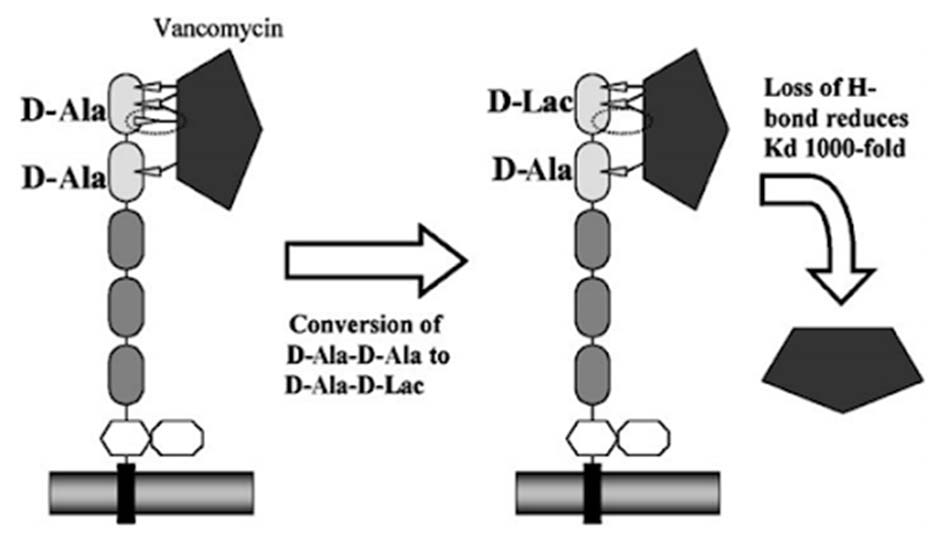
Cơ chế đề kháng chính của các vi khuẩn với kháng sinh nhóm này là thay đổi đích tác dụng. Vi khuẩn sẽ làm mọi cách để giảm ái lực gắn của kháng sinh với đầu D-Ala-D-Ala của lipid II. Một cơ chế đề kháng phổ biến là thay đổi đầu D-Ala-D-Ala thành D-Ala-D-Lac, từ đó ái lực liên kết với kháng sinh giảm 1000 lần do mất một liên kết hydro. Hoặc thay đổi đầu D-Ala-D-Ala thành D-Ala-D-Ser, ái lực liên kết giảm đi 6 lần do cản trở lập thể. Có 6 type đề kháng chính thuộc loại này, đó là VanA, VanB, VanC, VanD, VanE và VanG (ngoài ra sẽ còn một vài type khác). VanA và VanB được quy định trên plasmid và có thể truyền cho các vi khuẩn khác thông qua quá trình tiếp hợp, trong khi đó VanC, VanD, VanE và VanG được quy định trên nhiễm sắc thể.
Đề kháng kiểu VanA là cơ chế kháng thuốc hay gặp nhất ở các cầu khuẩn ruột Enterococcus. Chịu trách nhiệm cho đề kháng loại này là gen nhảy Tn1546 cùng một số yếu tố khác, chúng thay đổi cơ chất của kháng sinh từ D-Ala-D-Ala thành D-Ala-D-Lac. Đề kháng kiểu VanA của S.aureus được truyền lại từ Enterococcus.
Đề kháng kiểu VanB cũng được quy định thông qua gen nhảy (Tn1547 hoặc Tn1549), dẫn đến thay đổi đầu D-Ala-D-Ala thành D-Ala-D-Lac. Các chủng đề kháng kiểu VanB chỉ đề kháng mạnh với Vancomycin, mà không phải là Teicoplanin.
Đề kháng kiểu VanC được tìm thấy ở E.gallinarum, E.casseliflavus và E.flavescens, dẫn đến thay đổi đầu D-Ala-D-Ala thành D-Ala-D-Ser. Các vi khuẩn mang kiểu đề kháng này chỉ kháng với Vancomycin ở mức độ thấp và không kháng với các Glycopeptide khác.
Đề kháng kiểu VanD làm thay đổi đầu D-Ala-D-Ala thành D-Ala-D-Lac.
Đề kháng kiểu VanE khá tương đồng với VanC và là kiểu đề kháng nội tại của E.faecalis.
Đề kháng kiểu VanG dẫn đến thay đổi đầu D-Ala-D-Ala thành D-Ala-D-Ser và chỉ tạo ra đề kháng ở mức độ trung gian với Vancomycin.
Dược động học
Hấp thu: Tất cả các kháng sinh nhóm này đều có cấu trúc peptide với kích thước phân tử lớn nên không thể hấp thu qua đường tiêu hóa. Chúng thường được dùng theo đường tĩnh mạch hoặc tiêm bắp, đôi khi là đường uống (điều trị các nhiễm khuẩn trong lòng ống tiêu hóa).
Phân bố: Do bản chất thân nước nên khả năng phân bố của kháng sinh vào các mô và dịch trong cơ thể tốt, trừ dịch não tủy (chỉ qua được khi màng não bị viêm). Vancomycin có liên kết với protein huyết tương khoảng 50% và thể tích phân bố (Vd) 0.3-0.43 L/kg. Telavancin có liên kết với protein huyết tương khoảng 90% và Vd = 133-145 mL/kg. Liên kết với protein huyết tương của Dalbavancin (chủ yếu là albumin) là 93%. Oritavancin có liên kết với protein huyết tương khoảng 85% và Vd = 87.6 L.
Chuyển hóa: Thường không chuyển hóa hoặc chưa xác định được con đường chuyển hóa. Tuy nhiên với Dalbavancin, có phát hiện được một lượng nhỏ Hydroxy-Dalbavancin trong nước tiểu. Oritavancin ức chế yếu CYP2C9 và CYP2C19 của gan, đồng thời cảm ứng yếu CYP3A4 và CYP2D6.
Thải trừ: Các kháng sinh nhóm này thải trừ chủ yếu qua thận dưới dạng không đổi (khi dùng đường toàn thân) (trừ Dalbavancin chỉ thải trừ qua nước tiểu 33% dưới dạng không đổi và Oritavancin chỉ thải trừ qua nước tiểu dưới 5%) hoặc phân (khi dùng đường uống). Thời gian bán thải (t1/2) tăng lên ở bệnh nhân có bệnh thận.
t1/2 của Vancomycin là 4-6 giờ, Teicoplanin là 30-190 giờ, Telavancin là 8 giờ, Dalbavancin là 346 giờ (sau một liều đơn 1000 mg) và Oritavancin là 245 giờ ở bệnh nhân có chức năng thận bình thường.
Nghiên cứu và phát triển các kháng sinh Glycopeptide mới
Hiện nay, sự phát triển của các kháng sinh nhóm Glycopeptide mới đang đi theo hướng bán tổng hợp từ các kháng sinh có nguồn gốc tự nhiên là chủ yếu.
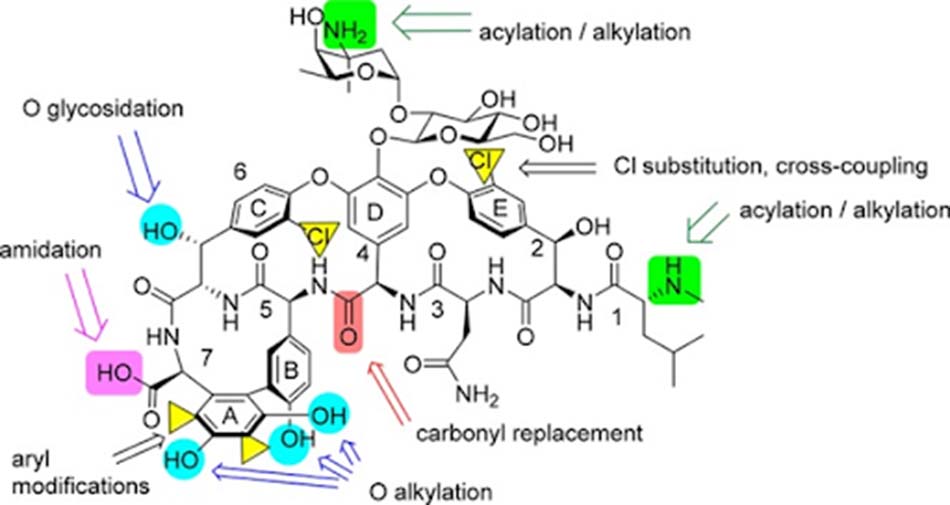
Chúng ta sẽ cùng đi đến một số phương pháp cụ thể đang được áp dụng để phát triển các phân tử kháng sinh nhóm Glycopeptide mới.
Tiếp cận nhắm mục tiêu màng
Đại học Queensland đã phát triển các phân tử gọi là Vancapticin, đây là các chất có cấu trúc tương tự Vancomycin, với đầu C được thay đổi bổ sung: Một nhóm linker bis-amine, một peptide tích điện dương EEPS (Electrostatic Effector Peptide Sequence) và một mũ kỵ nước MIE (Membrane Insertive Element). Các nhóm thế này có tác dụng hỗ trợ làm tăng nồng độ của phần khung Vancomycin trên bề mặt màng, đem đến cơ hội tương tác với lipid II cao hơn và đồng thời cũng tăng cường dimer hóa Vancomycin trên bề mặt màng. Với sự thay đổi này, hiệu lực chống lại MRSA của các kháng sinh mới tăng hơn 100 lần so với Vancomycin ban đầu. Ngoài cơ chế liên kết với lipid II và ức chế tổng hợp peptidoglycan như Vancomycin, các Vancapticin cũng đồng thời phá vỡ trực tiếp màng tế bào.
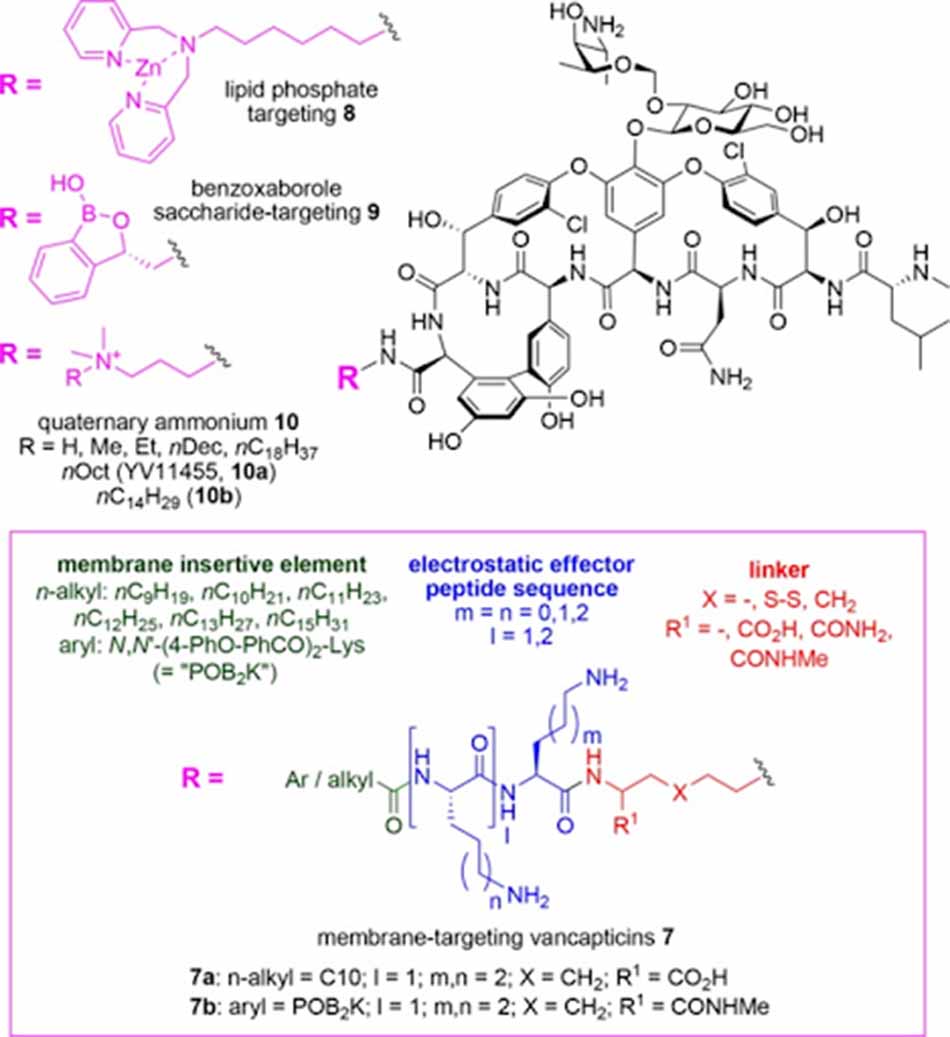
Thay đổi lõi Glycopeptide
Một cách tiếp cận khác để khắc phục tình trạng đề kháng với Vancomycin đã được nhóm Boger tại Viện Nghiên cứu Scripps mô tả năm 2006. Trong đó, nhóm carbonyl quan trọng của Vancomycin, tham gia tương tác với nhóm -NH- amide của lipid II Lys-D-Ala-D-Ala tripeptide, đã được thay thế. Như đã được để cập ở trên, vi khuẩn có thể đề kháng với Vancomycin thông qua đột biến thay đổi đầu Lys-D-Ala-D-Ala thành Lys-D-Ala-D-Lac, lúc này nguyên tử O trong nhóm ester của lactate không thể hình thành liên kết hydro với nhóm carbonyl của Vancomycin. Nhóm nghiên cứu công bố họ đã tổng hợp được phần aglycone của Vancomycin với nhóm carbonyl được thay thế bằng nhóm methylene. Aglycone mới này có ái lực liên kết với Lys-D-Ala-D-Ala giảm 35 lần, nhưng ái lực liên kết với Lys-D-Ala-D-Lac lại tăng 40 lần so với phần aglycone của phân tử Vancomycin ban đầu. Sau đó, các nhà khoa học đã tiếp tục tổng hợp các phần aglycone mới khác với nhóm carbonyl được thay thế bằng nhóm thioamide hoặc sau đó là amidine (2011). Dẫn chất thioamide mất tất cả hoạt tính liên kết, trong khi đó dẫn chất amidine có ái lực liên kết với Lys-D-Ala-D-Ala giảm chỉ 2 lần, đồng thời ái lực liên kết với Lys-D-Ala-D-Lac tăng tới 600 lần.
Năm 2014, aglycone amidine được glycosyl hóa đầy đủ đã được tổng hợp. Đồng thời, các nhà khoa học cũng phát hiện ra nhóm thế chlorobiphenyl ở đường vancosamine làm tăng hiệu lực trên VRE 100 lần. Cuối cùng, nhóm acid carboxylic được thay đổi năm 2017 bằng cách amide hóa với nhóm thế aminoalkylamine bậc 4. Thay đổi này giúp cho phân tử kháng sinh có thêm khả năng phá vỡ màng tế bào vi khuẩn (cơ chế bổ sung).
Thay đổi vòng thơm
Nhóm Boger đã kiểm tra lần lượt hoạt tính của Vancomycin khi thay các nhóm thế khác nhau vào vòng thơm E. Khi thay thế nhóm chloro trên vòng E bằng hydro hoặc các nhóm phân cực thì hoạt tính kháng sinh giảm, trong khi các nhóm thế không phân cực giữ nguyên hoạt tính. Methyl hóa nhóm hydroxy phenol hoặc carboxy acid làm tăng hoạt tính chống lại VanB E.faecalis 8 lần, nhưng hoạt tính trên tụ cầu vàng nhạy cảm Methicillin (VSSA: Vancomycin-Sensitive Staphylococcus aureus) giảm đi 2 lần. Cũng trên VSSA, loại bỏ nhóm thế chloro trên vòng C nhưng giữ lại trên vòng E làm giảm hoạt tính Vancomycin đi 8 lần, còn nếu làm ngược lại thì hoạt tính giảm đi 4 lần, nếu bỏ cả hai nhóm thế thì hoạt tính giảm đi 16 lần. Các sản phẩm bis-alkenyl hóa có hoạt tính giảm đi 32 lần. Nhóm Miller tại Đại học Yale cho thấy nếu như loại bỏ nhóm thế chloro trên vòng E, đồng thời sử dụng nhóm thế này trên vòng C để thực hiện phản ứng ghép cặp với các nhóm cồng kềnh thân lipid thì hoạt tính kháng sinh cũng giảm đi.
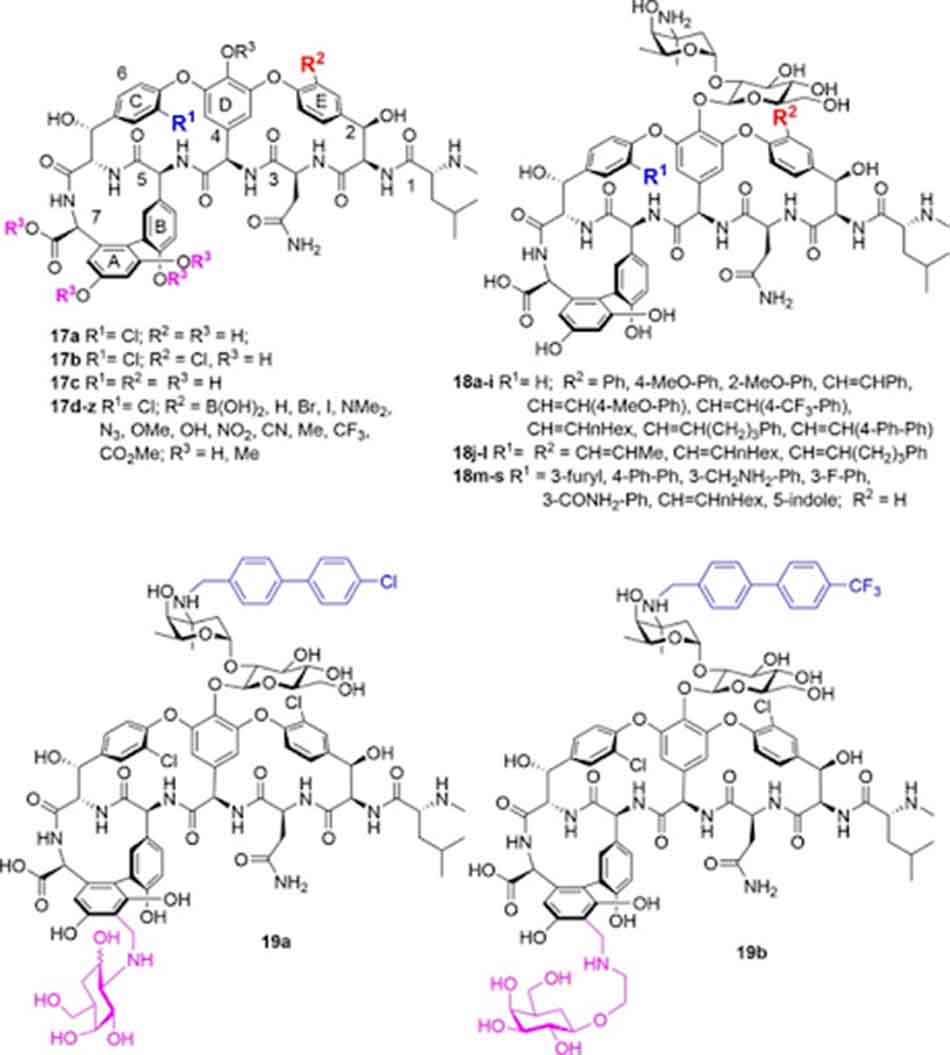
Các sản phẩm được bromo hóa có hoạt tính chống lại MRSA và E.faecalis loại VanA và VanB giảm đi 4 lần. Khi áp dụng chiến lược tương tự cho Teicoplanin, hoạt tính của kháng sinh cũng không có sự cải thiện đáng kể.
Năm 2018, một nhóm tại Thượng Hải đã tiến hành bổ sung thêm gốc đường vào vòng thơm thông qua nhóm aminomethyl (tương tự Telavancin), đồng thời alkyl hóa đường vancosamine (tương tự Oritavancin). Các hợp chất 19a và 19b (hình trên) là các hợp chất thể hiện hoạt tính tốt nhất.
Thay đổi ở nhóm hydroxyl
Nhóm Miller cũng đã sử dụng chiến lược phosphoryl hóa chọn lọc, với xúc tác đặc biệt để acyl hóa nhóm hydroxyl và bromo hóa vòng thơm của Vancomycin và Teicoplanin. Hoạt tính chống lại VanA E.faecalis được cải thiện một phần. Nhóm Boger đã tiến hành methyl hóa chọn lọc một hoặc nhiều nhóm phenol của phần aglycone của Vancomycin, tuy nhiên hoạt tính kháng sinh không đổi hoặc giảm đi.
Thay đổi ở đầu N
Phân tử Vancomycin có chứa nhóm N-methyl (thuộc Leu) ở đầu N. Desmethyl Vancomycin có hoạt tính khá tương đồng với Vancomycin và được sử dụng trên lâm sàng ở Trung Quốc từ năm 1967. Desmethyl Vancomycin được alkyl hóa chọn lọc trên nhóm amino của đường vancosamine bằng các nhóm thế thơm và kỵ nước có hoạt tính trên MRSA được cải thiện 2-4 lần. Dẫn chất phenylhexyl cũng cho thấy hoạt tính trên VRE tăng hơn 4 lần. Ngoài các thay đổi vừa được liệt kê, các thay đổi khác đều cho thấy sự cải thiện hoạt tính không đáng kể.
Dimer hóa Glycopeptide
Các Glycopeptide khi được dimer hóa cho thấy ái lực liên kết với lipid II mạnh hơn. Năm 1996, hai đầu C của Vancomycin đã được nối với nhau thông qua cầu nối alkyl, disulfide hoặc bisamine. Điều này làm giảm hoạt tính kháng sinh trên tụ cầu vàng, nhưng bù lại hoạt tính trên VRE tăng hơn 100 lần. Vài năm sau đó, các nhà khoa học tiếp tục dimer hóa Vancomycin thông qua các cầu nối disulfide hoặc methylene gắn vào nhóm amino của đường vancosamine. Chất có hoạt tính tốt nhất là 25e (n = 2) với hoạt tính trên MRSA tăng hơn 10 lần và trên VRE tăng hơn 100 lần. Ngoài cấu trúc dimer, cấu trúc trimer cũng đã được mô tả với cầu nối ở ba đầu C thông qua 1,3,5-tris(4-aminomethylanilinide)benzene.
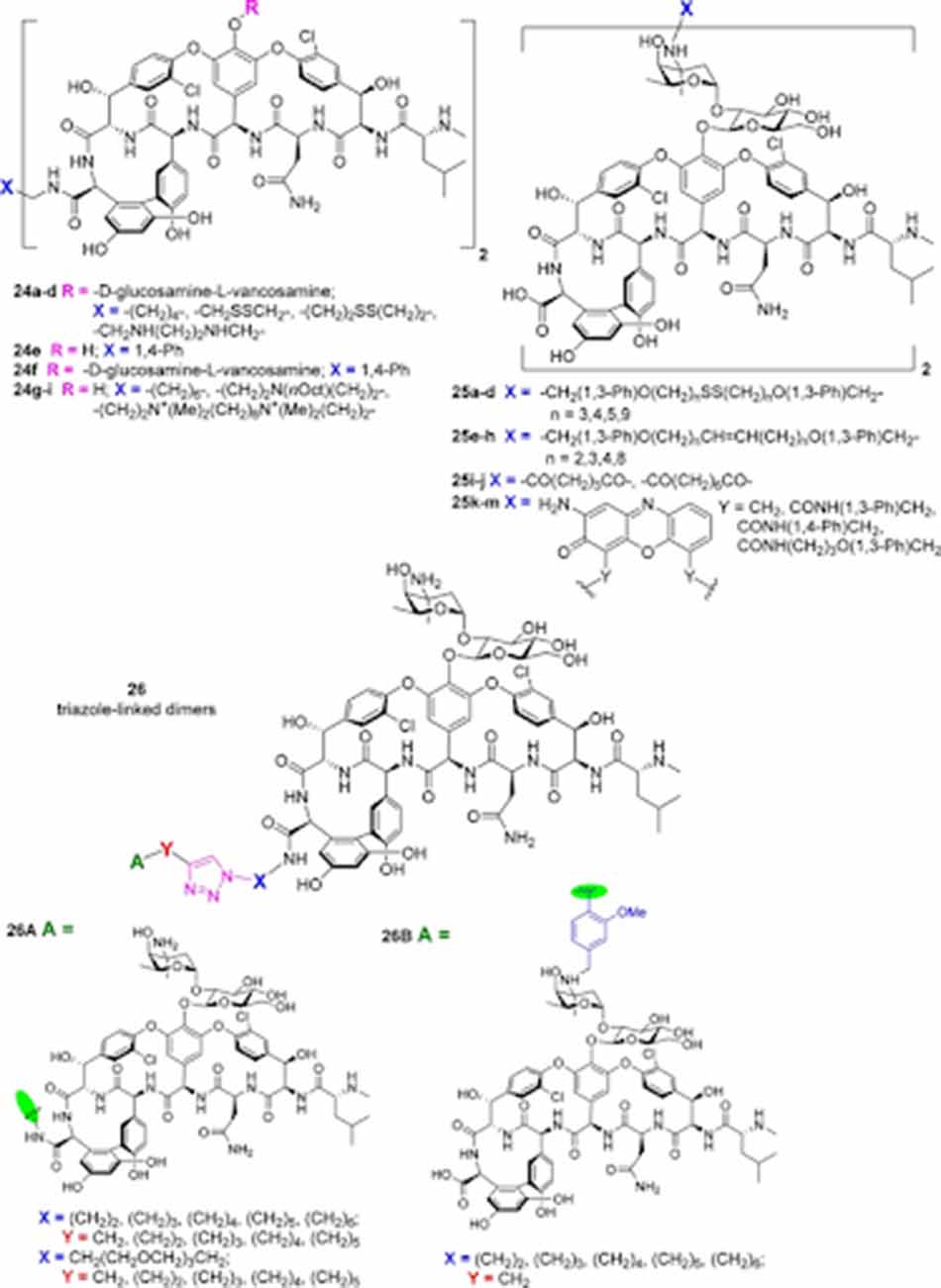
Vào năm 2003, Theravance đã tiến hành đánh giá 40 Glycopeptide dimer khác nhau, với các cầu nối thông qua đầu C (C), đầu N (N), nhóm amino của đường vancosamine (V) hoặc nhóm amine trên vòng thơm 7 (R). Các cặp sau đã được đánh giá: C-C, N-N, V-V, R-R, C-N, C-V, C-R, N-V, N-R và V-R. Cầu nối V-V với các liên kết ngắn thể hiện hoạt tính trên VanB VRE tốt nhất, tăng hơn 400 lần.
Các dimer 25k-m (hình trên) có hoạt tính trên MRSA/VRSA giảm 16-32 lần nhưng hoạt tính chống lại VRE tăng 8-32 lần.
Năm 2015, hai nhóm carboxy của hai aglycone của Vancomycin được nối với nhau qua cầu nối 1,8-diaminooctane, N,N-bis(3-aminopropyl)octylamine hoặc cấu nối chứa amine bậc 4 (24g-i trong hình trên). Các chất này có hoạt tính trên MRSA chỉ được cải thiện một chút, nhưng hoạt tính trên VRE tăng lên đáng kể.
Năm 2017, nhóm Sharpless đã nối hai hợp phần Vancomycin thông qua cầu nối triazole (26 trong hình trên). Hoạt tính trên VRE được cải thiện đáng kể.
Glycopeptide liên hợp
Vancomycin có thể được liên hợp với nhiều nhóm chức năng (các kháng sinh khác, nhóm siderophore hoặc fluorophore…). Ví dụ: Cefilavancin là phân tử kết hợp giữa Vancomycin và một Cephalosporin. Nó được phát triển đầu tiên bởi Theravance Biopharma, Inc., sau đó hợp tác với R-Pharm (10/2012) và tham gia thử nghiệm lâm sàng pha III (3/2015) về nhiễm trùng da và cấu trúc da cấp do vi khuẩn gram dương gây ra. Liên kết giữa hai phân tử kháng sinh này thông qua nhóm carboxy tại đầu C của Vancomycin và nhóm oxime của Cephalosporin. TD-1607 cũng là sự kết hợp giữa Vancomycin và một Cephalosporin, nhưng kiểu liên kết khác, thông qua nhóm aminomethyl trên vòng thơm 7 của Vancomycin với nhóm thế pyridinyl trên Cephalosporin. Nó đã được tham gia thử nghiệm lâm sàng pha I năm 2013, nhưng hiện nay đã bị ngừng phát triển.
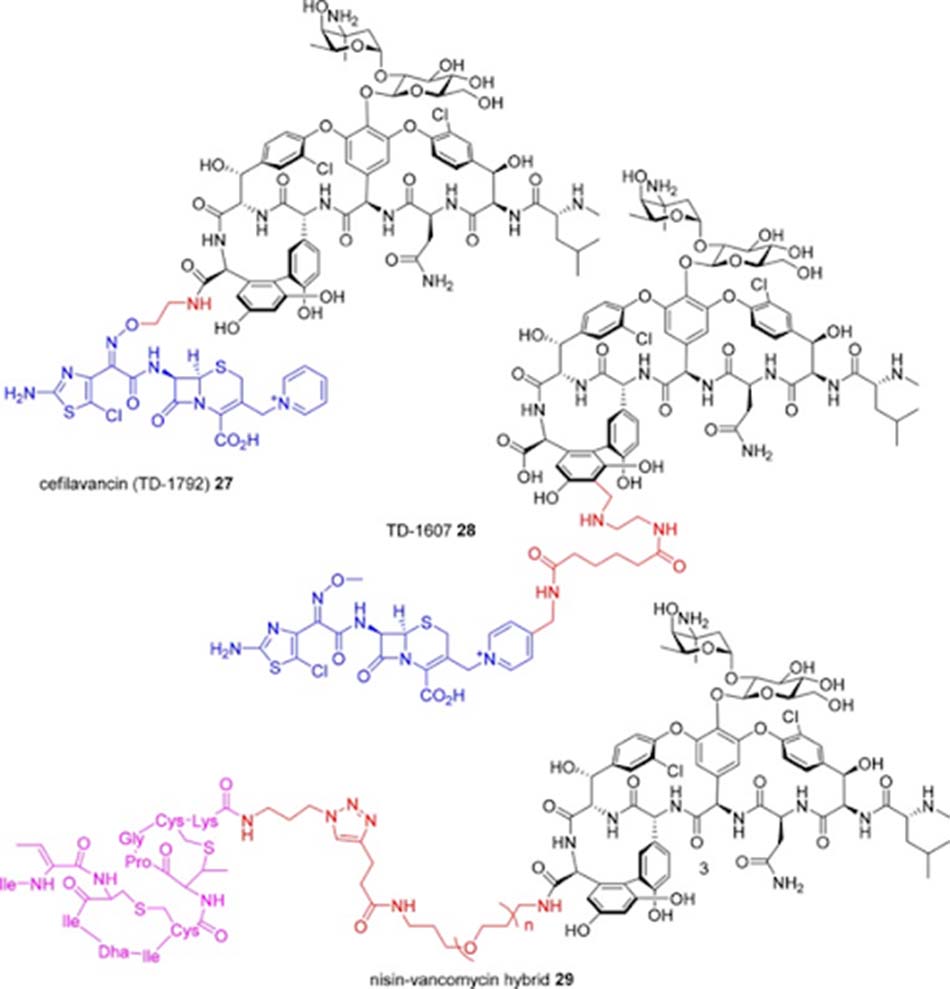
Vancomycin cũng từng được thử cho kết hợp với Nisin (nó liên kết với pyrophosphate của lipid II). Hoạt tính trên VRE của hợp chất này tăng khoảng 40 lần, với n = 3 (hợp chất 29 trong hình trên) được cho là hiệu quả nhất.
Các phối tử catechol và catechol/hydroxamate có khả năng liên kết với ion sắt, điều này làm cho vi khuẩn bắt buộc phải vận chuyển phức hợp kháng sinh – sắt vào bên trong tế bào. Chiến lược này được gọi là “Con ngựa thành Troy” và phối tử liên kết với Glycopeptide được gọi là “siderophore”. Trong khi đó, gốc dipicolyl gắn kẽm được gắn vào đầu C của Vancomycin để liên kết với pyrophosphate của lipid thành tế bào. Tương tự, phối tử chứa pyridinyl – bạc cũng được tạo ra nhằm tăng cường hoạt tính kháng khuẩn của Vancomycin. Nếu đầu C của Vancomycin được liên kết với nhóm methoxyphenylamide (có tích hợp cầu nối PEG [PolyEthylene Glycol]), khả năng gắn vào xương của kháng sinh tăng lên do nhóm này liên kết mạnh với hydroxyapatite, điều này thích hợp với các nhiễm trùng xương. Tuy nhiên, tổn thương trên thận cũng tăng lên.
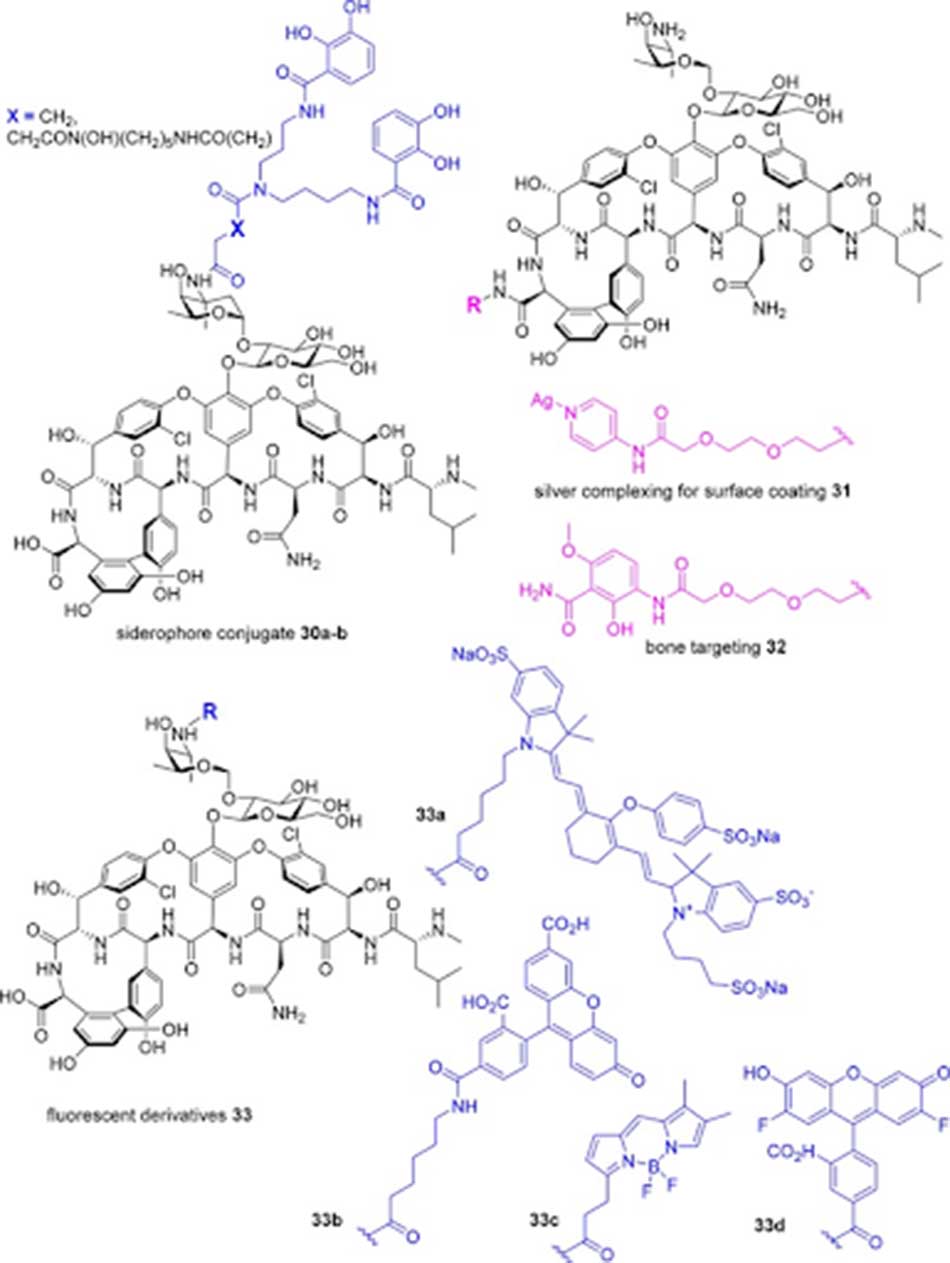
Tiền thuốc của Vancomycin có thể được tạo ra bằng cách gắn các nhóm PEG phân nhánh vào nhóm amine của đường vancosamine thông qua liên kết amide. Liên kết amide này sau đó sẽ bị thủy phân in vivo giải phóng Vancomycin.
Các hợp chất 33 trong hình trên là sự liên hợp của phân tử kháng sinh với các đầu dò huỳnh quang (các nhóm phân tử có khả năng phát huỳnh quang). Nhờ các đầu dò này mà chúng ta có thể theo dõi đường đi của kháng sinh trong cơ thể, đem lại lợi ích trong chẩn đoán và điều trị.
Sinh tổng hợp
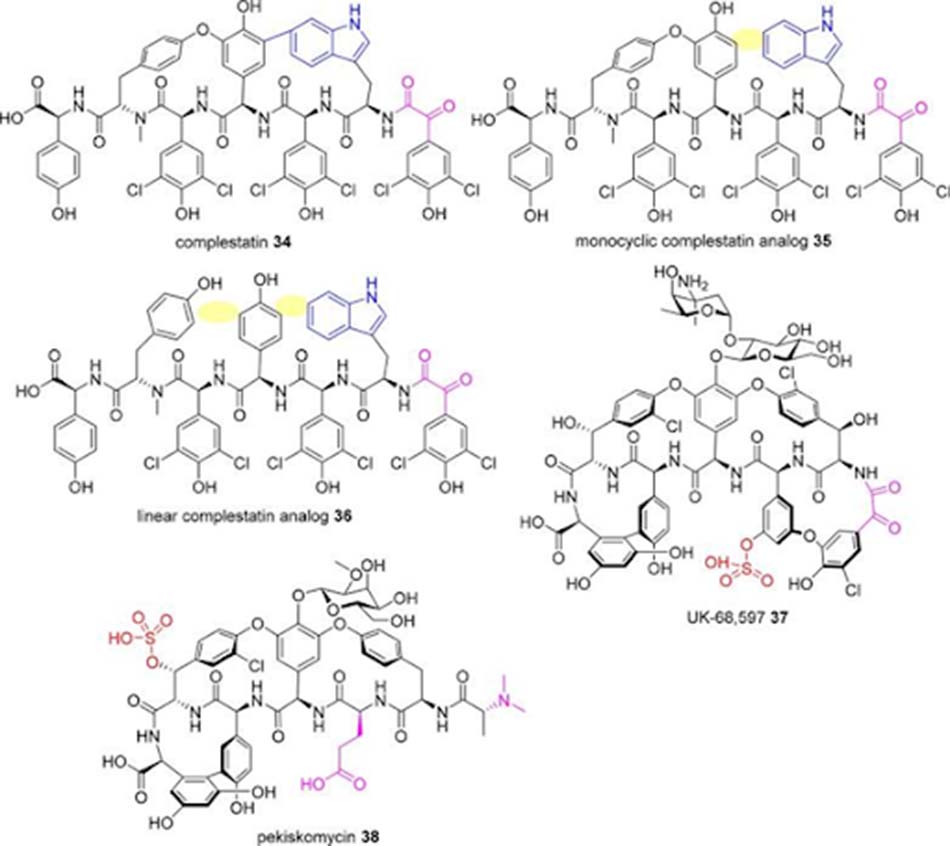
Complestatin là một aglycone của Glycopeptide type 5, được sinh tổng hợp từ Streptomyces lividans. UK-68,597 là một Glycopeptide tương tự Teicoplanin được sulfate hóa. Pekiskomycin là một Glycopeptide tự nhiên khác được ra đời từ chiến lược sàng lọc các cụm gen sinh tổng hợp mới kết hợp với bộ tiền lọc đề kháng Glycopeptide.
Tác dụng – Chỉ định của kháng sinh nhóm Glycopeptide
Các kháng sinh nhóm này đều có tác dụng diệt khuẩn, nhưng tốc độ diệt khuẩn và mô hình dược lực học/dược động học có nhiều sự khác biệt. Các Glycopeptide thế hệ thứ nhất như Vancomycin và Teicoplanin có tác dụng diệt khuẩn chậm, phụ thuộc thời gian. Các kháng sinh thế hệ hai lại có khả năng diệt khuẩn nhanh và mạnh hơn, đặc biệt Oritavancin cho thấy tác dụng diệt khuẩn phụ thuộc nồng độ.
Các chỉ định của từng loại kháng sinh như sau:
– Vancomycin: Viêm đại tràng do S.aureus (kể cả MRSA), tiêu chảy do C.difficile, viêm nội tâm mạc do S.gallolyticus, Corynebacterium và Enterococcus (kết hợp với Aminoside) hoặc bệnh nhân dị ứng với Penicillin, viêm nội tâm mạc van giả khởi phát sớm do S.epidermidis (kết hợp với Aminoside và Rifampin), nhiễm khuẩn huyết, nhiễm trùng da và cấu trúc da, nhiễm trùng xương hoặc nhiễm trùng đường hô hấp dưới do MRSA, tụ cầu coagulase âm tính hoặc bệnh nhân không thể sử dụng β-lactam (dị ứng, không dung nạp hoặc thất bại điều trị…), dự phòng trước phẫu thuật (tiêu hóa, sinh dục, tim, lồng ngực, động mạch, mở hộp sọ, thay khớp hoặc cắt cụt chi).
– Teicoplanin: Nhiễm trùng da và mô mềm có biến chứng, nhiễm trùng xương khớp, viêm phổi cộng đồng và viêm phổi bệnh viện, nhiễm trùng đường tiết niệu có biến chứng, viêm nội tâm mạc, viêm phúc mạc do thẩm phân phúc mạc liên tục ngoại trú, nhiễm khuẩn huyết do bất cứ nhiễm trùng nào vừa được nêu, điều trị thay thế cho tiêu chảy và viêm đại tràng do C.difficile. Trong một số trường hợp, cần xem xét kết hợp Teicoplanin với kháng sinh khác.
– Telavancin: Nhiễm trùng da và cấu trúc da do MRSA, MSSA (Methicillin-Susceptible Staphylococcus aureus: Tụ cầu vàng nhạy cảm với Methicillin), S.pyogenes, S.agalactiae, S.anginosus, S.intermedius, S.constellatus và E.faecalis (chủng nhạy cảm với Vancomycin), viêm phổi bệnh viện, bao gồm cả viêm phổi thở máy do S.aureus (bao gồm cả MRSA và VISA [Vancomycin-Intermediate Staphylococcus aureus: Tụ cầu vàng đề kháng trung gian với Vancomycin]).
– Dalbavancin: Nhiễm trùng da và cấu trúc da do MRSA, MSSA, S.pyogenes, S.agalactiae, S.anginosus, S.intermedius và S.constellatus.
– Oritavancin: Nhiễm trùng da và cấu trúc da do MRSA, MSSA, S.pyogenes, S.agalactiae, S.dysgalactiae, S.anginosus, S.intermedius, S.constellatus và E.faecalis (chủng nhạy cảm với Vancomycin).
Cách dùng – Liều dùng kháng sinh nhóm
– Vancomycin:
+ Viêm đại tràng do S.aureus: 0.5-2 g/ngày PO (đường uống) chia ra mỗi 6-8 giờ. Thời gian điều trị 7-10 ngày.
+ Tiêu chảy do C.difficile: 125 mg PO mỗi 6 giờ. Thời gian điều trị 10 ngày.
+ Viêm nội tâm mạc, nhiễm khuẩn huyết, nhiễm trùng da và cấu trúc da, nhiễm trùng xương và nhiễm trùng đường hô hấp dưới: 500 mg IV (tĩnh mạch) mỗi 6 giờ hoặc 1 g IV mỗi 12 giờ. Liều hàng ngày khởi đầu không được thấp hơn 15 mg/kg.
+ Dự phòng phẫu thuật: Phẫu thuật tiêu hóa và sinh dục: Truyền chậm 1 g IV trong hơn 1 giờ, bắt đầu 1-2 giờ trước rạch da; Phẫu thuật tim, lồng ngực, động mạch, mở hộp sọ, thay khớp hoặc cắt cụt chi: Truyền 15 mg/kg IV trong 1-2 giờ, bắt đầu trong vòng 2 giờ trước rạch da.
– Teicoplanin:
+ Nhiễm trùng da và mô mềm có biến chứng, nhiễm trùng đường tiết niệu có biến chứng và viêm phổi: Liều nạp 6 mg/kg IV hoặc IM (tiêm bắp) mỗi 12 giờ x 3 lần, liều duy trì 6 mg/kg IV hoặc IM mỗi 24 giờ. Nồng độ đáy trong huyết tương cần duy trì trên 15 mg/L.
+ Nhiễm trùng xương khớp: Liều nạp 12 mg/kg IV mỗi 12 giờ x 3-5 lần, liều duy trì 12 mg/kg IV hoặc IM mỗi 24 giờ. Nồng độ đáy trong huyết tương cần duy trì trên 20 mg/L.
+ Viêm nội tâm mạc: Liều nạp 12 mg/kg IV mỗi 12 giờ x 3-5 lần, liều duy trì 12 mg/kg IV hoặc IM mỗi 24 giờ. Nồng độ đáy trong huyết tương cần duy trì trên 30 mg/L. Thời gian điều trị tối thiểu 21 ngày và tối đa 4 tháng.
+ Tiêu chảy và viêm đại tràng do C.difficile: 100-200 mg PO x 2 lần/ngày. Thời gian điều trị 7-14 ngày.
+ Viêm phúc mạc do thẩm phân phúc mạc liên tục ngoại trú: Liều nạp 6 mg/kg IV x 1 lần, sau đó tuần thứ nhất 20 mg/L trong túi đựng dung dịch thẩm tách, tuần thứ hai 20 mg/L trong các túi khác nhau và tuần thứ ba 20 mg/L trong túi qua đêm.
– Telavancin: Liều cố định 10 mg/kg IV mỗi 24 giờ. Thời gian điều trị là 7-14 ngày với nhiễm trùng da và cấu trúc da, 7-21 ngày với viêm phổi bệnh viện.
– Dalbavancin: 1500 mg IV 1 liều duy nhất hoặc 1000 mg IV ban đầu sau đó 500 mg IV sau 1 tuần. Truyền IV trong hơn 30 phút.
– Oritavancin: 1200 mg IV 1 liều duy nhất truyền IV trong hơn 3 giờ.
Tác dụng không mong muốn
Khi sử dụng theo đường uống, các kháng sinh nhóm này có thể gây ra đau bụng, đầy hơi, buồn nôn và nôn, hạ kali máu.
Khi dùng theo đường toàn thân, các tác dụng không mong muốn sau đây có thể xảy ra:
– Phản ứng tiêm truyền: Kích ứng tại chỗ tiêm, đau và hoại tử nơi tiêm, viêm tĩnh mạch, sốt, ớn lạnh…
– Rối loạn tiêu hóa: Buồn nôn và nôn, tiêu chảy, xuất huyết tiêu hóa, đi ngoài phân đen…
– Rối loạn hệ miễn dịch: Hội chứng “người đỏ” (chủ yếu do truyền quá nhanh), phát ban, phù mạch, viêm da tróc vảy, hồng ban đa dạng, hội chứng Stevens-Johnson, hoại tử thượng bì nhiễm độc, hội chứng DRESS (Drug Reaction with Eosinophilia and Systemic Symptoms: Phản ứng thuốc với tăng bạch cầu ái toan và các triệu chứng toàn thân)…
– Tổn thương thận (tổn thương thận cấp, viêm thận kẽ) và tai (ù tai, mất thính giác, chóng mặt, rối loạn tiền đình).
– Rối loạn hệ tạo máu: Giảm bạch cầu, giảm tiểu cầu, tăng bạch cầu ái toan, thiếu máu bất sản.
– Rối loạn hệ thần kinh: Đau đầu, chóng mặt, co giật…
– Rối loạn hệ hô hấp: Co thắt phế quản gây khó thở, thở khò khè.
– Cận lâm sàng bất thường: Tăng men gan, tăng creatinine máu.
– Bội nhiễm nấm miệng, nấm âm đạo, viêm đại tràng giả mạc do C.difficile (Dalbavancin).
– Hạ đường huyết (Dalbavancin).
Chống chỉ định sử dụng kháng sinh nhóm Glycopeptide
Quá mẫn cảm với bất cứ thành phần nào của thuốc.
Chống chỉ định phối hợp Heparin không phân đoạn IV với Telavancin.
Chống chỉ định sử dụng Heparin không phân đoạn IV trong vòng 120 giờ sau khi sử dụng Oritavancin.
Tương tác thuốc
Phối hợp Vancomycin với các thuốc gây mê: Liên quan đến ban đỏ và phản ứng phản vệ.
Phối hợp với các thuốc có độc tính trên thận khác (Aminoside, Amphotericin B, Colistin, Cisplatin…): Tăng nguy cơ gặp phải độc tính trên thận. Thận trọng với phối hợp này và chú ý theo dõi chức năng thận thường xuyên.
Phối hợp Vancomycin hoặc Teicoplanin với các kháng sinh nhóm Aminoside hoặc kháng sinh ức chế màng tế bào (Colistin, Daptomycin): Tạo ra tác dụng hiệp đồng tăng mức (“1 + 1 > 2”), tăng cường tác dụng diệt khuẩn và giảm nguy cơ đề kháng thuốc, nhưng đồng thời cũng làm tăng chi phí điều trị, tăng nguy cơ tương tác thuốc và tăng độc tính trên thận. Các phối hợp này thường được sử dụng các nhiễm khuẩn bệnh viện, nhiễm khuẩn nặng và đe dọa tính mạng. Vancomycin hoặc Teicoplanin là các kháng sinh diệt khuẩn chậm và phụ thuộc thời gian, có hiệu quả cao khi số lượng vi khuẩn tại ổ nhiễm khuẩn ít, trong khi các kháng sinh nhóm Aminoside hoặc kháng sinh ức chế màng tế bào như Colistin và Daptomycin là các kháng sinh diệt khuẩn nhanh, mạnh và phụ thuộc nồng độ, số lượng vi khuẩn tại ổ nhiễm khuẩn càng cao thì kháng sinh càng hiệu quả. Dựa vào mô hình dược động học/dược lực học (PK/PD) của kháng sinh, chế độ liều của sự phối hợp kháng sinh này sẽ được tối ưu hóa.
Phối hợp Telavancin với các thuốc khác cũng gây kéo dài khoảng QT trên điện tâm đồ (ECG) (Kháng sinh nhóm Macrolide, thuốc chống trầm cảm ba vòng [TCAs], thuốc điều trị loạn nhịp tim nhóm I và III…): Tăng nguy cơ gặp phải xoắn đỉnh, rung thất và ngừng tim. Thận trọng với phối hợp này và nên theo dõi ECG thường xuyên.
Lưu ý và thận trọng khi sử dụng kháng sinh nhóm Glycopeptide
Cảnh báo
– Vancomycin: Các chế phẩm chứa tá dược PEG 400 và N-acetyl-D-alanine (NADA) có thể gây dị tật thai nhi trong các nghiên cứu trên động vật. Tránh sử dụng các chế phẩm chứa tá dược này cho phụ nữ có thai.
– Telavancin: Thuốc làm tăng tỷ lệ tử vong ở những bệnh nhân suy thận nặng và vừa (thanh thải creatinine ClCr ≤ 50 mL/phút) khi điều trị viêm phổi bệnh viện, bao gồm cả viêm phổi thở máy. Thuốc cũng gây suy thận hoặc làm nặng lên tình trạng suy thận, cần theo dõi chức năng thận ở tất cả các bệnh nhân. Tránh sử dụng thuốc cho phụ nữ có thai trừ khi lợi ích là vượt trội so với rủi ro.
Thận trọng khác
Truyền tĩnh mạch Vancomycin hoặc Teicoplanin quá nhanh có thể dẫn đến hội chứng “người đỏ”, với các triệu chứng mày đay, phát ban đỏ, ngứa, hạ huyết áp.
Các tổn thương trên thận tăng lên khi liều sử dụng đường toàn thân tăng, bệnh nhân đã bị tổn thương thận từ trước hoặc dùng các thuốc khác cũng độc với thận.
Chỉ sử dụng Vancomycin để dự phòng viêm nội tâm mạc cho những bệnh nhân có nguy cơ cao.
Độc tính trên tai tỷ lệ với lượng thuốc sử dụng đường toàn thân cũng như thời gian dùng thuốc.
Giảm bạch cầu là tác dụng không mong muốn có thể hồi phục sau khi ngừng thuốc.
Telavancin cần được truyền trong tối thiểu 1 giờ. Thuốc có thể gây kéo dài khoảng QT trên ECG.
Sử dụng Telavancin, Dalbavancin và Oritavancin có thể gây ra tiêu chảy do C.difficile.
Bệnh nhân được điều trị bằng Oritavancin được báo cáo bị viêm tủy xương nhiều hơn so với Vancomycin trong các thử nghiệm lâm sàng.
Telavancin và đặc biệt là Oritavancin gây ảnh hưởng đến các xét nghiệm đông máu: Kéo dài aPTT (activated Partial Thromboplastin Time: Thời gian thromboplastin hoạt hóa từng phần), PT (Prothrombin Time: Thời gian prothrombin), INR (International Normalized Ratio: Tỷ số bình thường hóa quốc tế) và ACT (Activated Clotting Time: Thời gian đông máu hoạt hóa). Tuy nhiên các thuốc này không làm ảnh hưởng đến hệ thống đông máu.
Sử dụng thuốc trên đối tượng đặc biệt
Bệnh nhân suy giảm chức năng gan thận: Cần đặc biệt thận trọng với bệnh nhân suy giảm chức năng thận, bởi các kháng sinh nhóm này hầu như đều có con đường thải trừ chính qua thận, thêm vào đó đa số chúng đều độc với thận. Cần hiệu chỉnh liều theo ClCr nếu cần thiết. Với bệnh nhân suy giảm chức năng gan, nhìn chung không cần hiệu chỉnh liều ở những bệnh nhân suy giảm chức năng gan nhẹ hoặc vừa, còn với bệnh nhân suy giảm chức năng gan nặng, cần xem xét thận trọng, bởi dù ít tham gia vào chuyển hóa thuốc, nhưng những bệnh nhân nặng như vậy thường có các thông số dược động học thay đổi mạnh (tăng Vd; giảm liên kết thuốc – protein huyết tương…).
Người già và trẻ em: Người già và trẻ em (đặc biệt là trẻ sơ sinh, trẻ sinh non thiếu tháng) là những đối tượng thường có chức năng thận kém hoặc chưa hoàn chỉnh, cũng cần hiệu chỉnh liều theo cân nặng hoặc ClCr khi cần thiết.
Phụ nữ có thai và đang cho con bú: Nhìn chung dữ liệu còn thiếu nhiều. Chỉ sử dụng thuốc trên các đối tượng bệnh nhân này khi thực sự cần thiết và lợi ích nhận được là vượt trội so với rủi ro.
Một số nghiên cứu và thử nghiệm lâm sàng
– Thử nghiệm lâm sàng đối chứng ngẫu nhiên so sánh Telavancin với Vancomycin trong điều trị viêm phổi bệnh viện do vi khuẩn gram dương đã đi đến kết luận: Telavancin không thua kém Vancomycin về đáp ứng lâm sàng trong điều trị viêm phổi bệnh viện do vi khuẩn gram dương.
– Một tổng quan hệ thống và phân tích gộp so sánh Linezolid với Vancomycin trong điều trị viêm phổi bệnh viện cho thấy: Linezolid và Vancomycin có hiệu lực và độ an toàn tương đương nhau trong điều trị viêm phổi bệnh viện.
– Thử nghiệm lâm sàng đối chứng ngẫu nhiên, đa trung tâm, mù đôi so sánh Linezolid với Teicoplanin trong điều trị nhiễm trùng do vi khuẩn gram dương ở bệnh nhân nặng đã cho kết luận: Linezolid và Teicoplanin có hiệu lực và độ an toàn tương đương trong điều trị nhiễm trùng do vi khuẩn gram dương ở bệnh nhân nặng. Linezolid có khả năng đào thải MRSA trong thời gian ngắn, chứng tỏ thuốc này có khả năng thâm nhập qua da và niêm mạc tốt hơn Teicoplanin.
– Nghiên cứu FAST 2 so sánh Telavancin với liệu pháp tiêu chuẩn trong điều trị nhiễm trùng da và cấu trúc da có biến chứng gây ra bởi vi khuẩn gram dương cho thấy: Nhìn chung, Telavancin có hiệu quả tương đương liệu pháp tiêu chuẩn trên tất cả các đối tượng nghiên cứu, bao gồm cả những bệnh nhân đã có MRSA từ thời điểm ban đầu.
– Thử lâm sàng đối chứng ngẫu nhiên so sánh hai chế độ liều của Dalbavancin, bao gồm chế độ 1 liều duy nhất và chế độ 2 liều đưa ra kết luận: Chế độ 1 liều duy nhất không thua kém chế độ 2 liều, có tính an toàn tương đương và tiện lợi hơn chế độ 2 liều.
– Thử nghiệm lâm sàng đối chứng ngẫu nhiên, nhãn mở đánh giá hiệu quả của Dalbavancin trong điều trị viêm tủy xương ở người trưởng thành đưa ra kết luận: Chế độ 2 liều của Dalbavancin có hiệu qua và dung nạp tốt trong điều trị viêm tủy xương ở người trưởng thành.
– Thử nghiệm lâm sàng đối chứng ngẫu nhiên, mù đôi đánh giá hiệu quả của Oritavancin 1 liều duy nhất trong điều trị nhiễm trùng da và cấu trúc da cấp do vi khuẩn cho kết luận: Oritavancin 1 liều duy nhất không thua kém Vancomycin 2 lần/ngày trong 7-10 ngày trong điều trị nhiễm trùng da và cấu trúc da cấp do vi khuẩn gram dương.
Tài liệu tham khảo
Mark A. T. Blaskovich, Karl A. Hansford, Mark S. Butler, ZhiGuang Jia, Alan E. Mark, and Matthew A. Cooper. “Developments in Glycopeptide Antibiotics”. ACS Infect Dis. 2018 May 11; 4(5): 715-735.
Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5952257/
Daina Zeng, Dmitri Debabov, Theresa L. Hartsell, Raul J. Cano, Stacy Adams, Jessica A. Schuyler, Ronald McMillan, and John L. Pace. “Approved Glycopeptide Antibacterial Drugs: Mechanism of Action and Resistance”. Cold Spring Harb Perspect Med. 2016 Dec; 6(12): a026989.
Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5131748/
Elisa Binda, Flavia Marinelli, and Giorgia Letizia Marcone. “Old and New Glycopeptide Antibiotics: Action and Resistance”. Antibiotics (Basel). 2014 Dec; 3(4): 572–594.
Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4790382/
Jeff Pootoolal, John Neu, Gerard D. Wright. “Glycopeptide antibiotic resistance”. Annu Rev Pharmacol Toxicol. 2002; 42: 381-408.
Ethan Rubinstein, Tahaniyat Lalani, G. Ralph Corey, Zeina A. Kanafani, Esteban C. Nannini, Marcelo G. Rocha, Galia Rahav, Michael S. Niederman, Marin H. Kollef, Andrew F. Shorr, Patrick C. Lee, Arnold L. Lentnek, Carlos M. Luna, Jean-Yves Fagon, Antoni Torres, Michael M. Kitt, Fredric C. Genter, Steven L. Barriere, H. David Friedland, Martin E. Stryjewski, and for the ATTAIN Study Group. “Telavancin versus Vancomycin for Hospital-Acquired Pneumonia due to Gram-positive Pathogens”. Clin Infect Dis. 2011 Jan 1; 52(1): 31–40.
Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3060890/
Andre C. Kalil, Michael Klompas, Gleb Haynatzki, and Mark E. Rupp. “Treatment of hospital-acquired pneumonia with linezolid or vancomycin: a systematic review and meta-analysis”. BMJ Open 2013; 3: e003912.
Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3808765/
Jorge A Cepeda, Tony Whitehouse, Ben Cooper, Janeane Hails, Karen Jones, Felicia Kwaku, Lee Taylor, Samantha Hayman, Steven Shaw, Christopher Kibbler, Robert Shulman, Mervyn Singer, A Peter R Wilson. “Linezolid versus teicoplanin in the treatment of Gram-positive infections in the critically ill: a randomized, double-blind, multicentre study”. J Antimicrob Chemother. 2004 Feb; 53(2): 345-55.
Available online: https://academic.oup.com/jac/article/53/2/345/850551
Martin E. Stryjewski, Vivian H. Chu, William D. O’Riordan, Brian L. Warren, Lala M. Dunbar, David M. Young, Marc Vallée, Vance G. Fowler Jr., Joel Morganroth, Steven L. Barriere, Michael M. Kitt, G. Ralph Corey, and for the FAST 2 Investigator Group. “Telavancin versus Standard Therapy for Treatment of Complicated Skin and Skin Structure Infections Caused by Gram-Positive Bacteria: FAST 2 Study”. Antimicrob Agents Chemother. 2006, 50(3): 862.
Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1426424/
Michael W Dunne, Sailaja Puttagunta, Philip Giordano, Dainis Krievins, Michael Zelasky, James Baldassarre. “A Randomized Clinical Trial of Single-Dose Versus Weekly Dalbavancin for Treatment of Acute Bacterial Skin and Skin Structure Infection”. Clin Infect Dis. 2016 Mar 1; 62(5): 545-51.
Available online: https://academic.oup.com/cid/article/62/5/545/2462963
Urania Rappo, Sailaja Puttagunta, Vadym Shevchenko, Alena Shevchenko, Alena Jandourek, Pedro L Gonzalez, Amy Suen, Veronica Mas Casullo, David Melnick, Rosa Miceli, Milan Kovacevic, Gertjan De Bock, Michael W Dunne. “Dalbavancin for the Treatment of Osteomyelitis in Adult Patients: A Randomized Clinical Trial of Efficacy and Safety”. Open Forum Infect Dis. 2018 Dec 10; 6(1): ofy331.
Available online: https://academic.oup.com/ofid/article/6/1/ofy331/5235615
Ralph Corey, M.D., Heidi Kabler, M.D., Purvi Mehra, M.D., Sandeep Gupta, M.D., J. Scott Overcash, M.D., Ashwin Porwal, M.D., Philip Giordano, M.D., Christopher Lucasti, M.D., Antonio Perez, M.D., Samantha Good, Ph.D., Hai Jiang, Ph.D., Greg Moeck, Ph.D., et al., for the SOLO I Investigators. “Single-Dose Oritavancin in the Treatment of Acute Bacterial Skin Infections”. N Engl J Med 2014; 370: 2180-2190.
Available online: https://www.nejm.org/doi/full/10.1056/nejmoa1310422
Tham khảo thêm:




{kind=link}