









![]()
Thành lập với sứ mệnh đưa thông tin y dược đến gần mọi người hơn.
Hotline: 0375.288.862
Địa chỉ: Khu đô thị Kim Văn – Kim Lũ, Đại Kim, Hoàng Mai, HN
Thuốc kháng sinh: Cơ chế tác dụng, phân loại 9 nhóm kháng sinh

Kháng sinh là gì?
Kháng sinh hiện nay không có gì xa lạ với nhiều người. Đây là một trong những loại thuốc được sử dụng nhiều nhất, cũng như cũng là một trong những loại thuốc bị lạm dụng và kê đơn sai nhiều nhất trên lâm sàng.
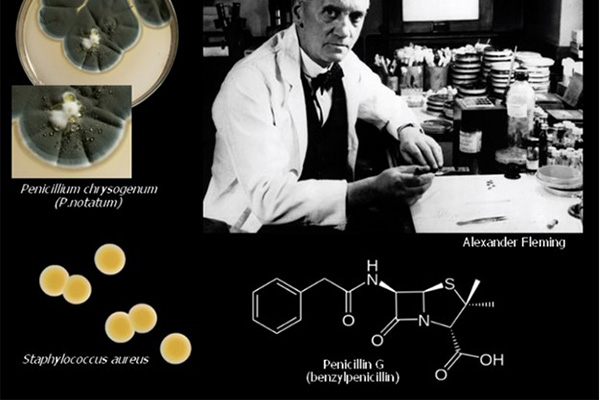
Kháng sinh đầu tiên được con người tìm là chính là Penicillin, một kháng sinh vô cùng nổi tiếng, và người đã tìm ra nó là bác sĩ Alexander Fleming, đồng thời cũng là một nhà sinh học và nhà nghiên cứu dược lý người Scotland. Sự kiện phát hiện ra Penicillin được coi là đã mở ra một kỉ nguyên mới của y học hiện đại. Kể từ đó, các kháng sinh mới liên tục được khám phá và phát triển, giúp đẩy lùi dần nhiều loại nhiễm trùng nguy hiểm.
Về định nghĩa kháng sinh, chúng ta có thể hiểu theo 2 nghĩa: Nghĩa rộng hoặc nghĩa hẹp. Trong cả 2 kiểu định nghĩa, kháng sinh đều là các hợp chất hóa học, có thể có nguồn gốc tự nhiên, tổng hợp hoặc bán tổng hợp, thường có phân tử lượng không lớn (từ nhỏ đến trung bình). Theo nghĩa hẹp, kháng sinh là các hợp chất có khả năng ức chế hoặc tiêu diệt vi khuẩn ở nồng độ nhỏ. Còn ở định nghĩa rộng, có thể hiểu kháng sinh không chỉ có thể ức chế hoặc tiêu diệt vi khuẩn, mà nó còn có tác dụng này trên các virus, nấm, động vật nguyên sinh và cả tế bào ung thư ở người. Tuy nhiên, trong thực tế lâm sàng, chúng ta thường hiểu từ kháng sinh theo nghĩa hẹp. Do đó trong bài viết này, chúng ta cũng chỉ sử dụng định nghĩa về kháng sinh theo nghĩa là “các hợp chất có khả năng chống lại vi khuẩn”.
(Bản thân từ kháng sinh trong tiếng Anh là “Antibiotics”, cũng có nghĩa là “chống lại sự sống”, do đó hiểu định nghĩa kháng sinh theo nghĩa rộng không hề sai. Do đó để phân biệt các kháng sinh theo nghĩa hẹp với các thuốc kháng nấm, virus, động vật nguyên sinh hay tế bào ung thư, đôi lúc người ta có thể dùng từ “Antibacterials”).
Tại sao kháng sinh lại quan trọng?
Kháng sinh quan trọng bởi nó là loại thuốc duy nhất có hiệu lực đủ mạnh để có thể sử dụng trong dự phòng hoặc điều trị nhiễm trùng do vi khuẩn.
Trước khi có kháng sinh, các trường hợp nhiễm trùng đều gần như không có thuốc chữa, chỉ có thể xảy ra 2 trường hợp: Hoặc là cơ thể người chiến thắng vi khuẩn, hoặc là vi khuẩn chiến thắng cơ thể người. Nếu cơ thể chúng ta chiến thắng vi khuẩn thì thường không có gì đáng nói, nhưng trường hợp thứ hai thường hay xảy ra hơn. Nếu ai đó “may mắn” thì vẫn có thể xử trí vết thương nhiễm trùng bằng cách cắt bỏ bộ phận nhiễm trùng (ví dụ như tay hoặc chân). Nhưng nếu như các vị trí nhiễm trùng là các trường hợp không thể cắt bỏ (ví dụ như viêm phổi, viêm thận – bể thận, viêm bàng quang…) thì bệnh nhân gần như chắc chắn sẽ tử vong. Các trường hợp phẫu thuật trước khi có kháng sinh ra đời thường có tỷ lệ tử vong rất cao. Kể cả là khi sau này đã áp dụng các biện pháp sát trùng bên ngoài, tỷ lệ tử vong đã giảm nhiều nhưng vẫn ở mức khá cao, nếu so với hiện tại.
Kể từ khi kháng sinh ra đời, kháng sinh đã trở thành “anh hùng” thực sự khi cứu sống hàng trăm triệu người trên khắp thế giới. Giờ đây chúng ta không còn lo lắng nhiều về nhiễm trùng như trước kia. Các trường hợp viêm phổi, viêm thận – bể thận, nhiễm trùng huyết hay thậm chí là viêm màng não nguy hiểm cũng gần như không còn đáng ngại. Thực hiện các phẫu thuật hiện nay cũng không còn lo lắng nhiều về nguy cơ tử vong hậu phẫu do nhiễm trùng nữa.
Như vậy, chúng ta có thể thấy kháng sinh chính là một trong những phát minh vĩ đại nhất và là bước ngoặt của lịch sử y khoa thế giới.
Vi khuẩn kháng kháng sinh
Vi khuẩn cũng là những sinh vật sống, và chúng luôn tìm cách thích nghi để tồn tại. Nói theo Thuyết Tiến hóa của Darwin, chúng sẽ tìm cách thay đổi chính mình, hay chính là tiến hóa để chống lại sự tấn công từ kháng sinh. Và sự thật đúng là như vậy. Hiện nay thi vi khuẩn kháng kháng sinh là một trong những vấn đề nhức nhối nhất trong y khoa và đã trở thành một vấn đề toàn cầu. Nó là nguyên nhân của ngày càng nhiều các ca tử vong trên thế giới. Các siêu vi khuẩn đề kháng kháng sinh (super bug) thậm chí có thể đề kháng lại tất cả các kháng sinh mà con người hiện có. Điều này làm cho việc điều trị nhiễm trùng ngày càng trở nên khó khăn hơn.
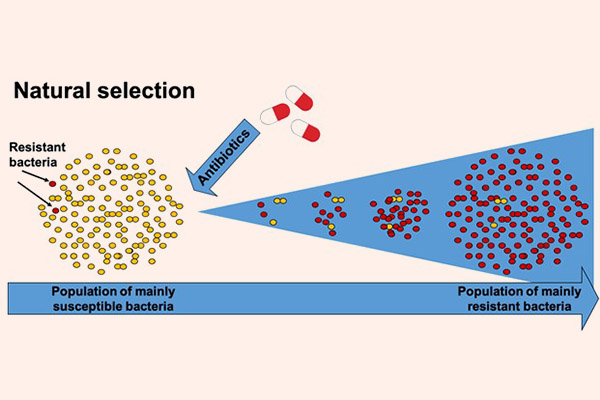
Sự đề kháng kháng sinh của vi khuẩn còn nguy hiểm ở chỗ: Các vi khuẩn có khả năng kháng chéo, có nghĩa là một khi chúng đã kháng một loại kháng sinh, thì chúng thường có xu hướng kháng thêm với các kháng sinh khác có cùng kiểu cấu trúc hóa học hoặc cùng cơ chế tác dụng. Điều này, cộng với việc nhiều vi khuẩn còn có khả năng trao đổi các gen kháng thuốc cho nhau, làm cho tình trạng kháng kháng sinh càng trở nên đáng sợ (1 đoạn gen kháng thuốc có thể quy định sự đề kháng nhiều loại thuốc cùng một lúc).
Có thể chống lại sự đề kháng kháng sinh của vi khuẩn không? Đáng tiếc rằng câu trả lời là không. Đó là quy luật của sự tiến hóa và cho dù loài người hay vi khuẩn thì cũng nằm trong quy luật đó. Muốn vi khuẩn không kháng kháng sinh thì chỉ có cách duy nhất là đừng sử dụng kháng sinh. Còn nếu đã sử dụng, thì vấn đề vi khuẩn kháng kháng sinh chỉ là vấn đề thời gian. Nhưng chúng ta có thể quyết định tốc độ đề kháng kháng sinh của vi khuẩn nhanh hay chậm, vì điều này phụ thuộc chủ yếu vào việc sử dụng kháng sinh có hợp lý hay không. Nhưng đáng tiếc là để đạt được điều này là rất khó, không chỉ ở các nước đang phát triển như Việt Nam, mà kể cả ở những nước phát triển như Hoa Kỳ, nơi đã phát hiện ra các siêu vi khuẩn kháng lại tất cả các kháng sinh hiện có.
Nguyên nhân chủ yếu của sự đề kháng kháng sinh là sử dụng kháng sinh không hợp lý, là sự lạm dụng kháng sinh. Hành vi kê đơn kháng sinh không hợp lý của bác sĩ, tự ý bán kháng sinh mà không có đơn thuốc của dược sĩ và việc bệnh nhân sử dụng kháng sinh không đúng cách (không đủ liều, không đủ thời gian…) là những nguyên nhân phổ biến nhất. Việc kê đơn kháng sinh không cần thiết của bác sĩ hoặc tự ý bán kháng sinh không cần đơn thuốc của dược sĩ giúp tăng thu nhập và lợi nhuận cho họ. Còn về phía bệnh nhân, chúng ta cũng gặp không ít các trường hợp bệnh nhân sau khi dùng kháng sinh một số ngày đầu, thấy bệnh gần như khỏi đã tự ý ngừng sử dụng thuốc, hoặc một số bệnh nhân lại quá lạm dụng kháng sinh, sử dụng kháng sinh ngay cả khi các bệnh lý của họ không cần thiết sử dụng đến loại thuốc này.
Ngoài ra, sử dụng kháng sinh nhiều trên vật nuôi cũng được cho là một nguyên nhân quan trọng dẫn đến chọn lọc các chủng vi khuẩn đề kháng kháng sinh ngoài cộng đồng. Mà chính vật nuôi lại là những đối tượng thường được sử dụng những kháng sinh mạnh và độc tính cao. Lấy đơn cử như Colistin (giải pháp cuối cùng cho các nhiễm trùng vi khuẩn gram âm đa kháng) được sử dụng rộng rãi trên lợn tại Trung Quốc, hay Ceftriaxone (một Cephalosporin thế hệ 3) được sử dụng trong chăn nuôi tôm tại nhiều địa phương trên Việt Nam, ngay cả khi động vật nuôi không bị nhiễm khuẩn. Kháng sinh bị lạm dụng bởi chúng giúp vật nuôi khỏe mạnh, không bị bệnh, từ đó ăn khỏe và tăng trọng tốt hơn.
Vậy nếu như bạn là một người sử dụng kháng sinh hợp lý, luôn tuân thủ phác đồ điều trị và chỉ sử dụng kháng sinh trong những trường hợp thật sự cần thiết, thì liệu bạn có nguy cơ nhiễm trùng vi khuẩn kháng kháng sinh hay không? Câu trả lời là có. Rất tiếc là cho dù bạn có sử dụng kháng sinh hợp lý như thế nào đi chăng nữa, thì bạn vẫn có thể bị nhiễm trùng vi khuẩn kháng kháng sinh. Vi khuẩn đó có thể là từ người khác, cũng có thể là từ nguồn chứa vi khuẩn kháng kháng sinh lớn, chính là các bệnh viện. Chống lại các vi khuẩn kháng kháng sinh thực sự không dễ, chỉ một vài cá nhân là không đủ, mà cần có sự chung tay của cả cộng đồng.
Như vậy các hãng dược lớn trên thế giới có giải pháp gì để đối phó với sự đề kháng kháng sinh hay không? Họ có thể tạo ra những kháng sinh mới chống lại các vi khuẩn đa kháng, giống như điều mà họ đã từng làm vào kỷ nguyên vàng của kháng sinh (những năm 1960, 1970), hay không? Lại một tin đáng buồn nữa là hầu hết các hàng dược lớn trên thế giới đã rút khỏi mảng nghiên cứu kháng sinh mới, do mảng này đem lại lợi nhuận không đáng kể. Chúng ta cũng không thể trách họ được, vì kháng sinh thực sự là một loại thuốc khó có thể đem lại nhiều lợi nhuận giống như các loại thuốc khác. Ví dụ: Các thuốc điều trị ung thư thường không được sử dụng nhiều (vì số bệnh nhân ung thư không nhiều), nhưng giá thành lại có thể rất đắt, nên thu được lợi nhuận cao. Các thuốc liên quan đến tim mạch, đái tháo đường, bệnh thận mạn tính… thì không đắt như các thuốc ung thư, nhưng người bệnh thường phải điều trị suốt đời, vậy nên cũng tạo ra lợi nhuận cao. Các thuốc điều trị giun sán thì không phổ biến, không được sử dụng nhiều và cũng không đắt đỏ, nhưng giun sán thì hầu như không có kháng thuốc, và chúng cũng thường không nguy hiểm, nên không mất công nghiên cứu các loại thuốc mới (các loại thuốc điều trị nhiễm giun sán hiện tại đã có từ khá lâu). Nhưng kháng sinh thì lại toàn mang những nhược điểm bất lợi: Giá thành thường không cao, thường chỉ sử dụng trong một thời gian ngắn (trừ một vài trường hợp đặc biệt), lại cộng thêm chỉ sử dụng được một thời gian là xuất hiện vi khuẩn kháng thuốc. Thêm vào đó, nếu một kháng sinh mới được ra đời mà mang những ưu điểm vượt trội chống lại vi khuẩn kháng thuốc, chúng sẽ ngay lập tức sẽ bị đưa vào danh sách các kháng sinh dự trữ và chỉ được sử dụng trong các trường hợp đặc biệt, như vậy thì loại kháng sinh đó sẽ không thể tiêu thụ nhiều được, điều này đồng nghĩa với việc các công ty dược sẽ không thể kiếm được nhiều lợi nhuận từ nó. Hiện nay, chỉ còn 4 công ty dược lớn trên thế giới là còn đầu tư cho nghiên cứu và phát triển kháng sinh mới, đó là Pfizer (Hoa Kỳ), Merck (Đức), Roche (Thụy Sĩ) và GSK (Anh), ngoài ra cũng còn một vài công ty nhỏ hơn khác.
Alexander Fleming, cha đẻ của Penicillin, có dự đoán trước được tương lai của kháng sinh sẽ như thế nào sau mấy chục năm ông qua đời (Alexander Fleming qua đời năm 1955) không? Có lẽ là không. Nhưng ông đã biết trước được vi khuẩn không sớm thì muộn cũng sẽ kháng thuốc, và ông hiểu được việc sử dụng kháng sinh hợp lý là một điều cực kỳ quan trọng. Trong một cuộc phỏng vấn với tờ The New York Times năm 1945, sau khi được trao giải Nobel Sinh lý và Y khoa nhờ phát hiện ra Penicillin, ông đã cảnh báo về việc lạm dụng kháng sinh có thể dẫn đến các chủng vi khuẩn kháng thuốc. Ông nói, “The thoughtless person playing with Penicillin treatment is morally responsible for the death of the man who succumbs to infection with the Penicillin-resistant organisms. I hope this evil can be averted.” Tạm dịch là, “Những người lạm dụng Penicillin ngày hôm nay phải chịu trách nhiệm đạo đức cho cái chết của những người nhiễm vi khuẩn kháng Penicillin sau này. Tôi hi vọng tai họa này có thể được đẩy lùi.”

Ngày 27/02/2017, WHO (Tổ chức Y tế Thế giới) đã công bố danh sách gồm 12 vi khuẩn kháng thuốc nguy hiểm, có thể trở thành mối đe dọa toàn cầu.
Ưu tiên 1: Ưu tiên quan trọng
- Acinetobacter baumannii kháng Carbapenem.
- Pseudomonas aeruginosa (trực khuẩn mủ xanh) kháng Carbapenem.
- Enterobacteriaceae (các trực khuẩn đường ruột gram âm, bao gồm Klebsiella, E.coli, Serratia và Proteus) kháng Carbapenem, sinh ESBL (β-lactamase phổ rộng).
Ưu tiên 2: Ưu tiên cao
- Enterococcus faecium kháng Vancomycin.
- Staphylococcus aureus (tụ cầu vàng) kháng Methicillin (MRSA), đề kháng trung gian hoặc kháng Vancomycin.
- Helicobacter pylori (vi khuẩn gây viêm loét dạ dày – tá tràng) kháng Clarithromycin.
- Campylobacter spp. kháng Fluoroquinolone.
- Salmonellae (gây bệnh thương hàn) kháng Fluoroquinolone.
- Neisseria gonorrhoeae (lậu cầu) kháng Cephalosporin và kháng Fluoroquinolone.
Ưu tiên 3: Ưu tiên trung bình
- Streptococcus pneumoniae (phế cầu) không nhạy cảm với Penicillin.
- Haemophilus influenzae kháng Ampicillin.
- Shigella spp. (gây bệnh lỵ) kháng Fluoroquinolone.
Cơ quan Quản lý Thực phẩm và Dược phẩm (FDA) Hoa Kỳ đã tạo ra nhiều ưu đãi cho các công ty phát triển kháng sinh. Các ưu đãi này có thể bao gồm ưu tiên phê duyệt nhanh, hay tăng thời gian tiếp thị độc quyền kháng sinh để công ty có thể thu thêm lợi nhuận…, nhưng có vẻ như tình hình cũng không khả quan hơn cho lắm.
Các cơ chế đề kháng kháng sinh của vi khuẩn
Phần này sẽ cung cấp cho bạn 5 cơ chế đề kháng kháng sinh đã được phát hiện trên lâm sàng. Mỗi cơ chế thường sẽ ứng với một vài loại kháng sinh khác nhau. Cụ thể như thế nào, chúng ta sẽ nói cụ thể khi đến từng phần riêng của mỗi kháng sinh. (Chú ý là cũng có thể có những cơ chế đề kháng kháng sinh khác không được đề cập đến ở đây, nhưng chúng không phổ biến)
- Cách 1: Thay đổi đích tác dụng của kháng sinh. Sự thay đổi này có thể là về số lượng hoặc ái lực gắn của kháng sinh với đích tác dụng. Cơ chế này có thể gặp với tất cả các nhóm kháng sinh. Nguyên nhân thường là do tự đột biến gen quy định tổng hợp đích tác dụng đó, hoặc lấy gen đột biến đó từ một vi khuẩn khác.
- Cách 2: Sản sinh ra enzyme làm bất hoạt kháng sinh. Các enzyme có thể là enzyme thủy phân, acetyl hóa, phosphoryl hóa… Cơ chế này đặc biệt nổi tiếng ở các vi khuẩn đề kháng với các kháng sinh thuộc nhóm β-lactam, chúng tiết ra một enzyme được gọi là các β-lactamase, có khả năng thủy phân vòng β-lactam trong cấu trúc phân tử kháng sinh. Các β-lactamase là các enzyme quan trọng bậc nhất và cũng được quan tâm nhiều nhất trên thực hành lâm sàng.
- Cách 3: Thay đổi tính thấm màng tế bào vi khuẩn với phân tử kháng sinh. Cơ chế này chỉ phổ biến ở các vi khuẩn gram âm. Để qua được màng tế bào vi khuẩn, kháng sinh thường phải khuếch tán qua các kênh protein đặc biệt xuyên màng tế bào, gọi là porin. Các vi khuẩn có thể thích nghi bằng cách giảm số lượng hoặc đột biến các kênh porin trên màng tế bào, làm kháng sinh khó có thể vào được bên trong tế bào vi khuẩn và thể hiện tác dụng của nó.
- Cách 4: Hình thành các bơm tống thuốc (Efflux Pump). Các bơm tống thuốc này có tác dụng đưa kháng sinh ra khỏi tế bào vi khuẩn khi chúng đã vào trong tế bào. Cơ chế này cũng chỉ phổ biến ở các vi khuẩn gram âm.
- Cách 5: Đi đường vòng, hay còn gọi là “bypass”. Trong trường hợp kháng sinh ức chế một chuỗi phản ứng đóng vai trò quan trọng với sự sống của tế bào vi khuẩn, vi khuẩn có thể thích nghi bằng cách “đi đường vòng”, có nghĩa là nó vẫn tổng hợp ra chất cần thiết cho mình, nhưng theo con đường khác, bỏ qua con đường cũ. Cơ chế này có thể xảy ra với nhóm kháng sinh ức chế tổng hợp acid folic: Các Sulfamides kháng khuẩn.
Các nguyên tắc sử dụng kháng sinh
Các nguyên tắc sử dụng kháng sinh được ra đời để hướng dẫn sử dụng kháng sinh hợp lý, nhằm tối ưu hóa sử dụng kháng sinh, tránh những lãng phí không cần thiết và hạn chế nguy cơ kháng thuốc đang tăng cao trong cộng đồng.
Chỉ sử dụng kháng sinh khi có nhiễm khuẩn
Điều mà tất cả mọi người cần đặc biệt nhớ, đó là kháng sinh chỉ có tác dụng điều trị các bệnh nhiễm khuẩn, nó không có tác dụng điều trị với các bệnh do nhiễm virus như cảm lạnh thông thường. Do đó, sử dụng kháng sinh trong các trường hợp không phải nhiễm khuẩn chỉ làm hao phí tiền bạc, tăng nguy cơ chọn lọc các chủng vi khuẩn đề kháng và còn có thể gặp phải các tác dụng không mong muốn.
Nếu như bác sĩ có trong tay kết quả vi sinh khẳng định nhiễm khuẩn, thì lúc đó lựa chọn sử dụng kháng sinh là hiển nhiên. Tuy nhiên, nếu như không có kết quả vi sinh trong tay, lúc này bác sĩ phải quyết định xem có điều trị bằng kháng sinh hay không. Nếu bệnh nhân có biểu hiện lâm sàng (và có thể là cả cận lâm sàng) của nhiễm khuẩn rõ ràng, thì lúc này bệnh nhân sẽ được kê đơn kháng sinh theo kinh nghiệm (Empiric Antibiotic). Nếu không, bác sĩ không nên kê đơn kháng sinh cho bệnh nhân.
Cảm lạnh, cảm cúm, viêm họng và viêm phế quản là những trường hợp điển hình của những bệnh lý hay gặp thường do virus gây ra. Đa phần những trường hợp này không cần sử dụng kháng sinh.
Lựa chọn kháng sinh phù hợp
Sau khi đã khẳng định bệnh nhân phải sử dụng kháng sinh, bước tiếp theo sẽ đến lựa chọn kháng sinh. Lựa chọn đúng kháng sinh thường dựa trên các cơ sở sau:
- Dược động học: Nồng độ kháng sinh tại cơ quan đích có đạt được ngưỡng mục tiêu để điều trị hay không? Đặc biệt là một số vị trí mà kháng sinh khó thấm đến như dịch não tủy, tuyến tiền liệt, xương… Kháng sinh thải trừ chủ yếu qua gan, thận cần chú ý bệnh nhân có suy giảm chức năng gan, thận hay không và có cần hiệu chỉnh liều không? Trường hợp nào thì dùng đường uống, trường hợp nào thì dùng đường tĩnh mạch, trường hợp nào nên dùng đường tại chỗ? Bệnh nhân là phụ nữ có thai hoặc phụ nữ cho con bú cần chú ý điều gì?…
- Dược lực học: Kháng sinh có phổ tác dụng trên vi khuẩn gây bệnh không? Tình trạng đề kháng trong khu vực (dịch tễ)? Nếu điều trị theo kinh nghiệm cần có thông tin về các vi khuẩn thường gây bệnh ở vị trí nhiễm khuẩn đó. Hoạt lực của kháng sinh đối với vi khuẩn như thế nào?…
- Cần chú ý đến cả điều kiện kinh tế của bệnh nhân mà lựa chọn kháng sinh cho phù hợp.
Sử dụng kháng sinh đúng và đủ
Đúng liều: Sử dụng kháng sinh đúng liều là rất quan trọng. Thiếu liều kháng sinh không những làm cho bệnh nhân lâu khỏi hoặc không thể khỏi được, mà còn làm tăng nguy cơ phát triển các chủng vi khuẩn đề kháng. Thừa liều kháng sinh cũng có thể làm tăng nguy cơ gặp phải các tác dụng không mong muốn cũng như độc tính. Tuy nhiên, quan điểm cần quán triệt ở đây là “Thừa còn hơn thiếu”. Nhiễm trùng mà không khỏi được thì rất nguy hại cho bệnh nhân và làm việc điều trị bệnh trở nên khó khăn hơn, trong khi đó, chưa có ai tử vong vì quá liều kháng sinh cả, đa số các kháng sinh là các thuốc có phạm vi điều trị rộng, quá liều một chút thường cũng không gây ra tác hại gì to lớn lắm so với việc không thể điều trị khỏi nhiễm khuẩn do dùng thiếu liều.
Đúng cách: Tuân thủ điều trị đóng một vai trò quan trọng quyết định đến thành bại của liệu pháp kháng sinh. Với đa số các kháng sinh thì việc sử dụng thuốc đều đặn theo giờ là rất quan trọng. Bác sĩ hoặc dược sĩ cần hướng dẫn cho bệnh nhân về cách dùng thuốc, thời gian dùng thuốc, dùng trước, trong hay sau ăn, lưu ý cách sử dụng với một số dạng bào chế đặc biệt…
Đủ thời gian: Một liệu trình kháng sinh thông thường kéo dài từ 7-10 ngày, một số phác đồ ngắn hơn chỉ từ 5-7 ngày. Sử dụng đủ thời gian giúp đảm bảo vi khuẩn đã được loại trừ hoàn toàn. Bệnh nhân rất nên tránh trường hợp khi sử dụng kháng sinh trong thời gian đầu, thấy bệnh đỡ đi nhiều, đã tự ý ngừng sử dụng thuốc. Điều này chỉ làm tăng thêm nguy cơ chọn lọc vi khuẩn đề kháng kháng sinh mà thôi, bởi thực tế khi này vi khuẩn chưa bị tiêu diệt hoàn toàn. Nếu không tiêu diệt nốt mà để cho số vi khuẩn còn lại có khả năng phục hồi được, khả năng cao là nó sẽ đề kháng với kháng sinh đó, làm cho việc điều trị trở nên khó khăn hơn.
Một số trường hợp đặc biệt có liệu trình điều trị khác bình thường:
- Viêm xương – tủy xương, viêm tuyến tiền liệt… (các vị trí mà kháng sinh khó thấm): Có thể cần điều trị từ vài tuần đến vài tháng.
- Lao: Cần điều trị tối thiểu từ 6-8 tháng.
- Nhiễm trùng H.pylori: Một đợt điều trị kéo dài từ 10-14 ngày.
- Bệnh lậu: Thường chỉ dùng kháng sinh 1 liều duy nhất (Khuyến nghị đầu tay của CDC [Trung tâm Kiểm soát và Phòng ngừa Dịch bệnh] Hoa Kỳ: Ceftriaxone 250 mg tiêm bắp 1 liều duy nhất + Azithromycin 1 g đường uống 1 liều duy nhất).
- Sử dụng Azithromycin trong nhiễm khuẩn hô hấp: Azithromycin là một kháng sinh có tính “hướng mô”, nó phân bố vào mô rất tốt và thải trừ rất chậm (thời gian bán thải t1/2 khoảng 2-4 ngày). Do đó, phác đồ điều trị thường ngắn hơn bình thường. Bệnh nhân thường được kê đơn Azithromycin 500 mg/ngày x 3 ngày hoặc Azithromycin 500 mg trong ngày đầu, từ ngày thứ hai trở đi 250 mg/ngày x 4 ngày (người lớn), hoặc 10 mg/kg/ngày x 3 ngày (trẻ em < 45 kg). Phác đồ này đảm bảo được nồng độ kháng sinh duy trì ở trên ngưỡng điều trị trong 5-7 ngày.
Tuy nhiên, với tình trạng kháng kháng sinh ở cộng đồng cao như ở Việt Nam hiện nay, liều dùng của Azithromycin có thể được xem xét tăng lên ở ngưỡng 500 mg/ngày x 5 ngày (người lớn), hoặc 12 mg/kg/ngày x 5 ngày (trẻ em < 45 kg).
Phối hợp kháng sinh hợp lý
Phối hợp kháng sinh trên lâm sàng là không dễ. Ưu điểm của nó là giúp mở rộng phổ tác dụng, tận dụng sự hiệp đồng tác dụng giữa các kháng sinh, tăng hiệu quả điều trị và giảm nguy cơ phát triển các chủng vi khuẩn đề kháng (không được phép tự ý giảm liều mỗi thuốc). Nhược điểm của nó là làm tăng chi phí cho người bệnh, tăng khả năng gặp các tác dụng không mong muốn cũng như tương tác thuốc và có nguy cơ phối hợp không đúng sẽ dẫn đến đối kháng tác dụng, làm giảm hiệu quả điều trị.
Phối hợp kháng sinh trên lâm sàng cần rất cẩn trọng và cân nhắc kỹ lưỡng. Các phối hợp kinh điển trên lâm sàng bao gồm:
- Phối hợp một β-lactam (hoặc kháng sinh ức chế tổng hợp thành tế bào khác như Vancomycin) với một Aminoside: Tạo ra tác dụng hiệp đồng tăng cường (“1+1 > 2”). Các nhà khoa học cho rằng các β-lactam ức chế tổng hợp thành tế bào vi khuẩn, tạo điều kiện cho các Aminoside xâm nhập vào trong tế bào và ức chế tiểu đơn vị ribosome 30S của vi khuẩn, từ đó ức chế tổng hợp protein của vi khuẩn, tạo ra tác dụng hiệp đồng tăng cường. Thêm vào đó, β-lactam là kháng sinh diệt khuẩn phụ thuộc thời gian, thường dùng nhiều lần trong ngày, số lượng vi khuẩn trong ổ nhiễm khuẩn càng ít thì tác dụng diệt khuẩn của nó càng tốt. Trái lại, Aminoside là kháng sinh diệt khuẩn phụ thuộc nồng độ, thường dùng 1 lần trong ngày, số lượng vi khuẩn trong ổ nhiễm khuẩn càng cao thì nó diệt được càng nhiều. Aminoside liều cao đầu tiên sẽ tiêu diệt mạnh vi khuẩn, làm giảm nhanh số lượng vi khuẩn đang rất đông trong ổ nhiễm khuẩn. Sau đó, đến lượt mình, các β-lactam sẽ tiêu diệt nốt số vi khuẩn còn lại trong ổ nhiễm khuẩn, vốn chỉ còn không nhiều.
- Phối hợp Trimethoprim với Sulfamethoxazole: Hai kháng sinh này ức chế 2 enzyme xúc tác cho 2 phản ứng khác nhau trong con đường tổng hợp acid folic của vi khuẩn. Mỗi kháng sinh này nếu dùng đơn độc chỉ có tác dụng kìm khuẩn, nhưng phối hợp 2 kháng sinh này với nhau lại tạo ra tác dụng diệt khuẩn. Phối hợp với tỷ lệ chuẩn là 1 phần Trimethoprim : 5 phần Sulfamethoxazole. 2 dạng hàm lượng thường gặp với dạng thuốc kết hợp này là 80 mg Trimethoprim + 400 mg Sulfamethoxazole, hoặc liều gấp đôi 160 mg Trimethoprim + 800 mg Sulfamethoxazole.
- Phối hợp một β-lactam với một chất ức chế β-lactamase: Tuy không phải là phối hợp 2 kháng sinh, nhưng đây là phối hợp rất kinh điển, có tác dụng tăng tiềm lực của kháng sinh dùng cùng, giúp kháng sinh lấy lại phổ vốn có của nó trên các vi khuẩn mà trước đây kháng sinh mất hiệu lực vì vi khuẩn sản xuất β-lactamase. Các chất ức chế β-lactamase cụ thể sẽ được giới thiệu ở dưới.
- Phối hợp thuốc trong điều trị lao: Đây là phối hợp bắt buộc do vi khuẩn lao rất dễ kháng thuốc cũng như rất khó điều trị. Điều trị tuyến đầu gồm 5 thuốc: S (Streptomycin), R (Rifampin hoặc các Ryfamycin khác), H (Isoniazid), Z (Pyrazinamide) và E (Ethambutol). Nhiều nước trên thế giới đã loại Streptomycin ra khỏi tuyến đầu và đưa nó vào danh sách các thuốc hàng thứ hai trong điều trị lao.
Một số phối hợp kháng sinh khác có thể gặp trên lâm sàng:
- Phối hợp một β-lactam với một Fluoroquinolone: Phối hợp này thường mang ý nghĩa mở rộng phổ tác dụng là chính.
- Phối hợp một β-lactam với một Macrolide hoặc Doxycycline: Phối hợp này cũng mang ý nghĩa mở rộng phổ tác dụng là chính, thường được sử dụng trong nhiễm khuẩn hô hấp. Thông thường người ta ít khi phối hợp một kháng sinh diệt khuẩn với một kháng sinh kìm khuẩn, vì một thuốc hoạt động tốt khi vi khuẩn đang nhân lên, nhưng thuốc còn lại lại kìm hãm sự phát triển của vi khuẩn đó. Nhưng đó chỉ là lý thuyết. Trên thực tế, ta vẫn có một vài phối hợp như thế này trên lâm sàng.
Dự phòng kháng sinh hợp lý
Dự phòng kháng sinh giúp giảm thiểu nguy cơ có thể nhiễm trùng ở một số bệnh nhân đặc biệt (bệnh nhân khi này chưa có nhiễm trùng). Dự phòng kháng sinh này đa phần là tuân theo phác đồ có sẵn, chỉ giới hạn trong một số trường hợp nhất định như: Trước khi chuẩn bị phẫu thuật, trước thủ thuật nha khoa ở bệnh nhân có van tim nhân tạo, bệnh nhân bị viêm đường hô hấp cấp do virus và có nguy cơ trở nặng (như trẻ em)…
Trong đợt đại dịch SARS 2003, việc sử dụng kháng sinh dự phòng cho các bệnh nhân đã nhiễm virus SARS tại Việt Nam, mặc dù đi ngược lại với quan điểm của WHO lúc đó, nhưng đã làm giảm rõ rệt tỷ lệ tử vong của bệnh nhân.
Tổng quan về các nhóm thuốc kháng sinh
Kháng sinh nhóm β-lactam
β-lactam là nhóm kháng sinh được phát hiện ra đầu tiên trên thế giới, với đại diện nổi tiếng chính là Penicillin G. Các kháng sinh nhóm này đều có cấu trúc vòng β-lactam trong phân tử rất đặc trưng.
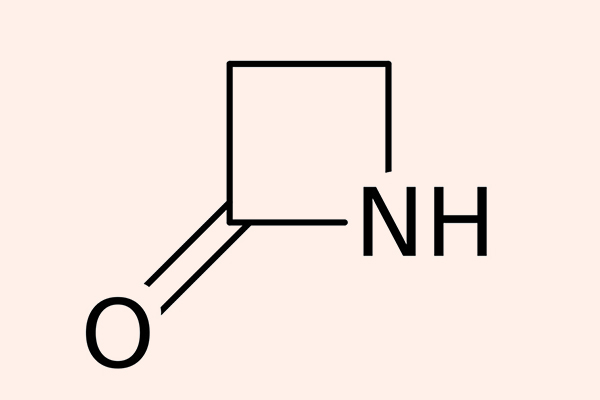
β-lactam là nhóm kháng sinh quan trọng nhất trên lâm sàng. Nói vậy bởi tần suất sử dụng nhóm kháng sinh này trên lâm sàng rất cao, phải đến hơn 60%. Thêm vào đó, số lượng kháng sinh trong nhóm này là cực kỳ lớn, với nhiều loại thuốc có những đặc tính dược động học cũng như dược lực học cực kỳ đa dạng. Do vậy, nhóm kháng sinh này cung cấp những thuốc để lựa chọn cho điều trị rất phong phú và đa dạng, gần như bất cứ nhiễm khuẩn ở bất kỳ cơ quan nào cũng có thể lựa chọn 1 kháng sinh nhóm này.
Cơ chế tác dụng
Cơ chế tác dụng của tất cả các β-lactam đều giống nhau, nên sẽ được trình bày ở phần chung này.
Tất cả các β-lactam đều là kháng sinh diệt khuẩn theo cơ chế ức chế tổng hợp thành tế bào. Thành tế bào vi khuẩn là một lớp nằm ngoài màng tế bào, có tác dụng duy trì hình dạng tế bào và bảo vệ cho tế bào khỏi bị vỡ bởi chênh lệch áp suất thẩm thấu. Các β-lactam đều ức chế tổng hợp thành tế bào thông qua ức chế tổng hợp peptidoglycan (một thành phần quan trọng trong thành tế bào) ở bước cuối cùng của quá trình sinh tổng hợp. Peptidoglycan là một thành phần rất vững chắc và bảo vệ vi khuẩn rất tốt. Cấu trúc phần peptidoglycan ở vi khuẩn gram dương và vi khuẩn gram âm có sự khác biệt: Lớp peptidoglycan ở thành tế bào vi khuẩn gram dương rất dày, trong khi lớp peptidoglycan ở thành tế bào vi khuẩn gram âm rất mỏng, thêm vào đó còn một lớp màng lipid ngoài cùng với nhiều thành phần phức tạo khác. Cá biệt các vi khuẩn nội bào (vi khuẩn không điển hình) không có thành tế bào nên chúng đề kháng tự nhiên với các β-lactam.
Mỗi đơn phân hình thành nên peptidoglycan gồm các thành phần sau: N-acetylglucosamine, N-acetylmuramic acid và 1 chuỗi pentapeptide (được gắn vào N-acetylmuramic acid). Đầu cuối của chuỗi pentapeptide là các acid amin D-alanyl-D-alanine (gọi tắt là D-Ala-D-Ala). Đích tác dụng của các kháng sinh nhóm β-lactam là enzyme transpeptidase, nhưng còn có một tên gọi khác là protein gắn Penicillin (Penicillin Binding Protein, kí hiệu là PBP). PBP chịu trách nhiệm cho sự cắt bỏ acid amin Alanine cuối cùng trong chuỗi pentapeptide, điều này dẫn đến sự hình thành liên kết chéo với một pentapeptide gần đó. Liên kết chéo làm cho thành tế bào trở nên bền vững hơn. Các kháng sinh nhóm β-lactam có cấu trúc hóa học tương đồng với D-Ala-D-Ala ở đầu pentapeptide, nên nó ức chế cạnh tranh với D-Ala-D-Ala trong liên kết với trung tâm hoạt động của PBP. Sự liên kết này làm ức chế phản ứng được xúc tác bình thường bởi transpeptidase. Khi phản ứng này bị ức chế, peptidoglycan không được tổng hợp do các liên kết chéo không được hình thành và dẫn đến là vi khuẩn chết. Nguyên nhân chi tiết tại sao vi khuẩn lại chết khi thành tế bào không được tổng hợp đầy đủ vẫn chưa được hiểu rõ ràng một cách hoàn toàn, nhưng có thể là do sự chênh lệch áp suất thẩm thấu giữa 2 bên thành tế bào đã làm tế bào vi khuẩn bị ly giải. Các kháng sinh nhóm β-lactam chỉ tiêu diệt vi khuẩn khi chúng đang ở trong giai đoạn phát triển, có sự tổng hợp tích cực thành tế bào. Với các vi khuẩn đang ở trạng thái nghỉ, chúng hầu như không có tác dụng.
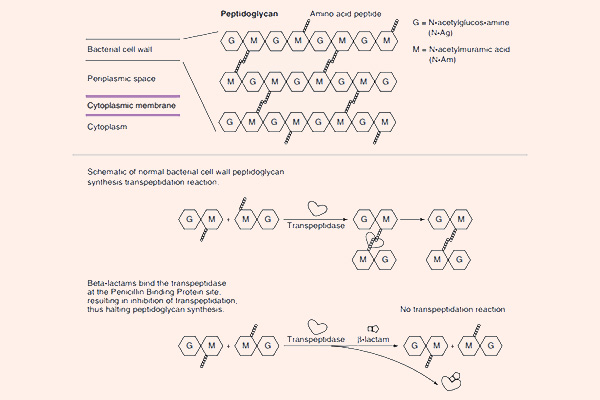
Cơ chế đề kháng
Các vi khuẩn có thể đề kháng với kháng sinh nhóm β-lactam theo 4 cơ chế:
- Sinh enzyme phá hủy thuốc β-lactamase: Đây là cơ chế đề kháng với kháng sinh nhóm β-lactam phổ biến nhất. Các nhà nghiên cứu đã tìm ra hàng trăm loại β-lactamase khác nhau. Các β-lactamase khác nhau rất nhiều về hoạt lực và phổ tác dụng. β-lactamase của các vi khuẩn như tụ cầu vàng, H.influenzae và E.coli có phổ khá hẹp, thường chỉ ưu tiên kháng sinh nhóm Penicillin là chính, thường không có tác dụng với các kháng sinh nhóm Cephalosporin. Nhưng có một số β-lactamase khác có phổ tác dụng rất rộng, như AmpC β-lactamase của trực khuẩn mủ xanh và Enterobacter sp. và các ESBL được sản xuất bởi các chủng Enterobacteriaceae, chúng mạnh tới mức có thể thủy phân cả vòng β-lactam của Cephalosporin, một nhóm kháng sinh có vòng β-lactam bền hơn Penicillin rất nhiều. Ngay cả các Carbapenem, các kháng sinh nhóm β-lactam bền vững nhất với β-lactamase, cũng không thể tránh khỏi số phận bi đát này. Các Carbapenem tuy rằng rất bền vững trước sự tấn công của các penicillinase và cephalosporinase, nhưng các enzyme carbapenemase và metallo-β-lactamase lại có thể thủy phân chúng dễ dàng.
Các vi khuẩn gram dương sinh β-lactamase tiết enzyme vào môi trường xung quanh nó, trong khi các vi khuẩn gram âm lại lưu giữ enzyme ở vùng không gian chu chất giữa màng tế bào và thành tế bào, vậy nên cơ chế sinh enzyme phá hủy thuốc này ở các vi khuẩn gram âm ưu việt hơn các vi khuẩn gram dương.
- Đột biến thay đổi cấu trúc của PBP, đích tác dụng của kháng sinh: Cơ chế đề kháng thuốc theo kiểu này thường xảy ra ở tụ cầu kháng Methicillin (MRSA hoặc MRSE), phế cầu kháng Penicillin và hầu hết các Enterococci kháng đa thuốc. Một khi đã kháng theo cơ chế này, chúng gần như sẽ kháng toàn bộ các kháng sinh nhóm β-lactam. Trường hợp đặc biệt là các Cephalosporin thế hệ 5 đã được thiết kế để tăng cường ái lực gắn của nó với PBP2a, giúp cho nó có thể điều trị được một số nhiễm trùng do các chủng tụ cầu kháng Methicillin.
- Giảm tính thấm của màng tế bào vi khuẩn với thuốc: Cơ chế này chỉ xảy ra ở vi khuẩn gram âm. Các tế bào vi khuẩn thích nghi với kháng sinh bằng cách giảm số lượng các kênh porin cho thuốc đi xuyên qua màng tế bào, hoặc xóa bỏ hoàn toàn các kênh này. Nhưng nếu chỉ có một mình cơ chế này thì vi khuẩn cũng rất khó kháng thuốc. Tuy nhiên, vùng không gian chu chất của các vi khuẩn gram âm này thường chứa β-lactamase, chịu trách nhiệm thủy phân thuốc ngay khi thuốc vừa vào tế bào. Điều này tạo nên cơ chế kháng thuốc kết hợp hiệu quả.
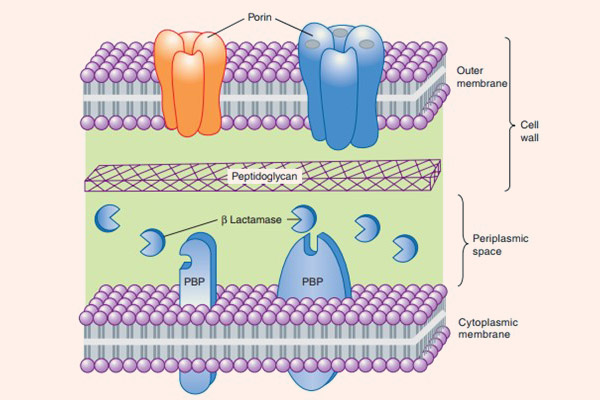
- Hình thành các bơm tống thuốc: Cơ chế này cũng chỉ có ở vi khuẩn gram âm, có tác dụng đẩy kháng sinh ra khỏi tế bào ngay khi chúng vừa vào không gian chu chất. Như vậy kháng sinh không thể tiếp cận được với mục tiêu của nó là PBP.
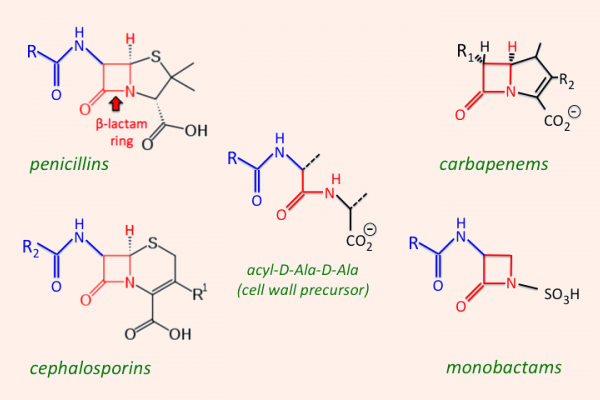
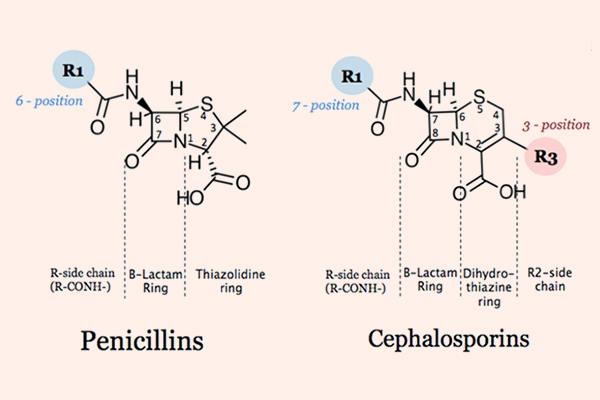
Các kháng sinh nhóm β-lactam được chia thành 4 nhóm lớn, với cấu trúc, các đặc tính dược động học và dược lực học có nhiều khác biệt, đó là: Penicillin, Cephalosporin, Carbapenem và Monobactam.
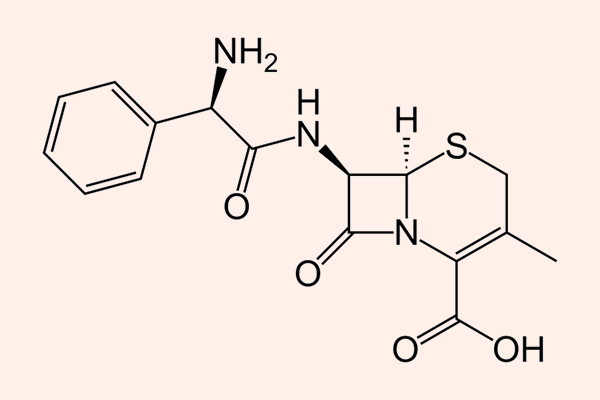
Penicillin
Penicillin G là kháng sinh được tìm thấy đầu tiên trên thế giới, và công lao tìm ra kháng sinh này thuộc về Alexander Fleming. Sự khám phá ra nó thực sự khá tình cờ, khi mà ông (hoặc ai đó) đã quên không làm sạch những đĩa petri đang nghiên cứu trước khi vào kì nghỉ dài. Sau khi hết kì nghỉ và trở lại phòng thí nghiệm, ông đã nhận thấy trong các đĩa petri này có mọc lên một loại vi sinh vật, và xung quanh loại vi sinh vật này, vi khuẩn không thể phát triển được, tạo thành một vùng không có vi khuẩn (chính là “vòng vô khuẩn”). Các nghiên cứu sâu hơn sau đó của ông đã chứng minh được rằng các vi sinh vật lạ này tiết ra một loại chất hóa học có tác dụng tiêu diệt vi khuẩn xung quanh nó. Sau này, chất hóa học đó được đặt tên là Penicillin và khi bắt đầu được sử dụng trong Chiến tranh Thế giới lần thứ hai (WWII), nó đã cứu sống rất nhiều binh sĩ bị thương và nhiễm trùng.
Hiện nay Penicillin là một thuật ngữ dùng để chỉ chung các kháng sinh có cùng cấu trúc khung phân tử giống Penicillin. Các Penicillin đều có cấu trung khung hóa học đặc trưng.
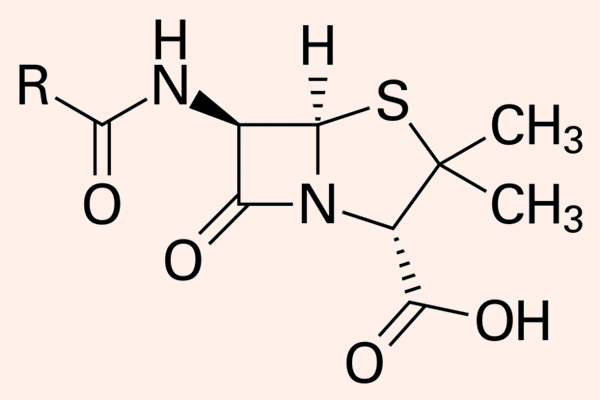
Dược động học
Đường dùng đa dạng (uống, tiêm truyền tĩnh mạch, tiêm bắp), chú ý rằng một kháng sinh có uống được hay không còn phụ thuộc vào sự ổn định trong acid dịch vị và khả năng hấp thu theo đường uống. Lưu ý một kháng sinh bền trong acid dịch vị thì chưa chắc đã uống được, nhưng nếu uống được thì chắc chắn nó bền trong acid dịch vị (không tính đến các dạng bào chế đặc biệt).
Sinh khả dụng thay đổi giữa các kháng sinh khác nhau, đa số chúng đều có quá trình hấp thu bị ảnh hưởng bởi thức ăn (trừ Amoxicillin), vậy nên hầu hết kháng sinh nhóm này phải dùng 1-2 giờ trước ăn. Dù sinh khả dụng của Amoxicillin không bị ảnh hưởng bởi thức ăn, nhưng đa phần các bác sĩ lâm sàng vẫn kê đơn chúng dùng trước bữa ăn.
Liên kết của kháng sinh với protein huyết tương cũng khác nhau giữa các Penicillin. Các kháng sinh phân nhóm này phân bố tốt vào các dịch trong cơ thể do có bản chất phân tử kháng sinh là thân nước. Thuốc có vào được sữa mẹ, nhưng ít vào được tuyến tiền liệt và hệ thần kinh trung ương (do khó qua hàng rào máu não), nhưng liều cao vẫn vào được dịch não tủy một cách tương đối và có thể điều trị một số trường hợp viêm màng não do một số chủng vi khuẩn nhạy cảm.
Con đường thải trừ chính của các kháng sinh phân nhóm này là nước tiểu, trừ Oxacillin, Cloxacillin và Dicloxacillin (các Penicillin chống tụ cầu) được thải trừ qua cả nước tiểu và mật, các thuốc này không cần hiệu chỉnh liều các thuốc này ở những bệnh nhân bị suy giảm chức năng thận (các thuốc còn lại thì cần). Ngoài ra các trường hợp bệnh nhân là trẻ sơ sinh có chức năng gan thận ở trẻ chưa được hoàn chỉnh cũng cần hiệu chỉnh liều theo cân nặng.
Tác dụng không mong muốn
Dị ứng và quá mẫn: Đây là tác dụng không mong muốn phổ biến nhất của các kháng sinh nhóm Penicillin. Các Penicillin có bản chất là những bán kháng nguyên, khi liên kết với protein huyết tương, chúng có thể trở thành các kháng nguyên hoàn chỉnh và hoạt hóa hệ miễn dịch. Các phản ứng dị ứng có thể nhẹ (ngứa, nổi mẩn, phát ban…), nhưng cũng có thể nặng và nguy hiểm tính mạng (phản ứng phản vệ, hội chứng Stevens-Johnson, hội chứng Lyell…). Bệnh nhân trước khi được cho sử dụng Penicillin nên được khai thác kĩ tiền sử dị ứng thuốc và làm test dị ứng trên da nếu cần thiết để loại trừ quá mẫn type 1. Nếu test âm tính, người bệnh có thể sử dụng Penicillin.
Các rối loạn tiêu hóa: Buồn nôn và nôn, tiêu chảy. Các thuốc đều có khả năng gây viêm đại tràng giả mạc do bùng phát Clostridium difficile, nhưng hay gặp nhất là với Ampicillin.
Người ta thường chia các Penicillin thành các phân nhóm nhỏ hơn như sau:
- Penicillin tự nhiên.
- Penicillin chống tụ cầu.
- Các Penicillin phổ rộng: Bao gồm các Aminopenicillin và Penicillin chống trực khuẩn mủ xanh.
Penicillin tự nhiên
Đại diện tiêu biểu cho nhóm này là Penicillin G và Penicillin V.
Các kháng sinh thuộc phân nhóm này có phổ tác dụng chủ yếu trên các vi khuẩn gram dương, cầu khuẩn gram âm và các vi khuẩn kị khí không sinh β-lactamase. Chúng không tiêu diệt được các trực khuẩn gram âm. Các Penicillin tự nhiên đều rất dễ bị phá hủy bằng β-lactamase. Đáng chú là liều của các Penicillin tự nhiên thường được tính theo đơn vị (Unit), chứ không tính theo khối lượng (mg) như nhiều kháng sinh khác.
Penicillin G được sử dụng theo đường tiêm, còn Penicillin V có thể uống được (do sự khác biệt trong độ bền với acid dịch vị).
Penicillin G là được chỉ định cho nhiễm trùng do liên cầu (viêm họng, nhiễm trùng da và mô mềm), não mô cầu (viêm màng não), phế cầu nhạy cảm với Penicillin (hiện tại tỷ lệ đề kháng Penicillin của phế cầu cao nên hiếm khi sử dụng Penicillin nhóm này để điều trị nhiễm khuẩn đường hô hấp do phế cầu), tụ cầu không sinh β-lactamase (nhiễm trùng da và mô mềm), Treponema pallidum (giang mai)…
Penicillin V có chỉ định tương tự Penicillin G. Thuốc có sinh khả dụng kém và phải dùng tới 4 lần/ngày, gây bất tiện cho người sử dụng thuốc. Hiện nay người ta thường ưa dùng Amoxicillin hơn.
Benzathine Penicillin và Procain Penicillin G được sử dụng theo đường tiêm bắp, với tác dụng chậm nhưng kéo dài. Chỉ định cho viêm họng do liên cầu tan huyết β nhóm A (1 liều duy nhất Benzathine Penicillin 1.2 triệu đơn vị), giang mai (liều Benzathine Penicillin cao hơn 2.4 triệu đơn vị 1 lần/tuần x 1-3 tuần), hoặc dự phòng thấp tim sau nhiễm liên cầu (cùng dùng 1 liều duy nhất Benzathine Penicillin).
Penicillin chống tụ cầu
Đại diện tiêu biểu cho nhóm này là Cloxacillin, Nafcillin, Oxacillin…
Các kháng sinh nhóm này có khả năng chống lại tụ cầu sinh β-lactamase do cấu trúc ở nhóm thế R1 cồng kềnh và có khả năng che chắn không gian bảo vệ vòng β-lactam. Chúng có phổ tác dụng trên các tụ cầu, liên cầu nhưng không có tác dụng trên các cầu khuẩn ruột, vi khuẩn kị khí, cầu khuẩn và trực khuẩn gram âm. Như vậy các kháng sinh nhóm này có phổ tác dụng hẹp hơn các Penicillin tự nhiên.
Thường chỉ định cho nhiễm trùng tụ cầu sinh β-lactamase nhạy cảm với Methicillin, chủ yếu trong nhiễm trùng da và mô mềm. Tụ cầu kháng Methicillin đề kháng với các kháng sinh nhóm này do đột biến thay đổi PBP2a (đích tác dụng của kháng sinh), làm giảm ái lực gắn của kháng sinh với PBP. Thuốc có thể được lựa chọn trong các trường hợp nhiễm trùng phế cầu kháng Penicillin nhưng nhạy cảm với Methicillin (mặc dù các trường hợp như thế này không nhiều).
Methicillin là Penicillin chống tụ cầu đầu tiên được phát triển và sử dụng trên lâm sàng, nhưng hiện tại đã không còn được sử dụng nữa do độc tính trên thận (gây viêm thận kẽ).
Penicillin phổ rộng
Đại diện tiêu biểu cho nhóm này là:
- Các Aminopenicillin: Amoxicillin và Ampicillin.
- Các Penicillin chống trực khuẩn mủ xanh: Carbenicillin, Ticarcillin, Piperacillin…
Phổ tác dụng của các kháng sinh nhóm này kém hơn về phía vi khuẩn gram dương so với Penicillin tự nhiên, nhưng phổ tác dụng về phía vi khuẩn gram âm lại được mở rộng hơn rất nhiều, thậm chí bao gồm cả trực khuẩn mủ xanh, vi khuẩn vốn kháng thuốc rất mạnh. Giống như các Penicillin tự nhiên, chúng cũng rất nhạy cảm với các β-lactamase, do đó khi sử dụng trên lâm sàng, chúng thường được phối hợp với các chất ức chế β-lactamase.
Amoxicillin và Ampicillin có cấu trúc gần giống nhau (chỉ khác nhau ở 1 nhóm -OH phenol), nhưng hấp thu qua đường uống của Amoxicillin lại vượt trội hơn hẳn. Amoxicillin thường được sử dụng 2 lần/ngày, còn Ampicillin lại lên tới 4 lần/ngày. Do đó khi sử dụng đường uống, Amoxicillin có ưu thế hơn hẳn so với Ampicillin về độ tiện lợi và khả năng duy trì tuân thủ điều trị ở bệnh nhân.
Amoxicillin thường được chỉ định cho nhiễm trùng hô hấp cả trên và dưới: Viêm mũi xoang, viêm tai giữa, viêm phế quản, viêm phổi mắc phải tại cộng đồng.
Ampicillin được chỉ định trong bệnh lỵ do nhiễm khuẩn Shigella. Phổ tác dụng của Ampicillin bao gồm các vi khuẩn kị khí, cầu khuẩn ruột, L.monocytogenes và các chủng cầu khuẩn và trực khuẩn gram âm không sinh β-lactamase như E.coli và Salmonella sp. Không sử dụng Ampicillin trong điều trị kháng sinh theo kinh nghiệm cho nhiễm trùng đường tiết niệu và bệnh thương hàn do tỷ lệ kháng thuốc đang ngày càng cao.
Các Penicillin chống trực khuẩn mủ xanh có thể được chia thành phân 2 loại nhỏ hơn: Carboxypenicillin và Ureidopenicillin. Đại diện cho các Carboxypenicillin là Carbenicillin và Ticarcillin. Đại diện cho Ureidopenicillin là Piperacillin. Ngoài trực khuẩn mủ xanh, chúng cũng tác dụng tốt trên nhiều vi khuẩn gram âm khác. Để chống lại sự đề kháng kháng sinh của trực khuẩn mủ xanh, chúng thường được phối hợp trong điều trị kháng sinh theo kinh nghiệm với các Aminoside hoặc Fluoroquinolone, đặc biệt là trong nhiễm trùng đường tiết niệu.
Ampicillin, Amoxicillin, Ticarcillin và Piperacillin đều được phối hợp với các chất ức chế β-lactamase: Acid clavulanic, Sulbactam hoặc Tazobactam. Các ví dụ về biệt dược nổi tiếng: Augmentin (Amoxicillin/Acid clavulanic), Unasyn (Ampicillin/Sulbactam), Timentin (Ticarcillin/Acid clavulanic) và Zosyn (Piperacillin/Tazobactam).
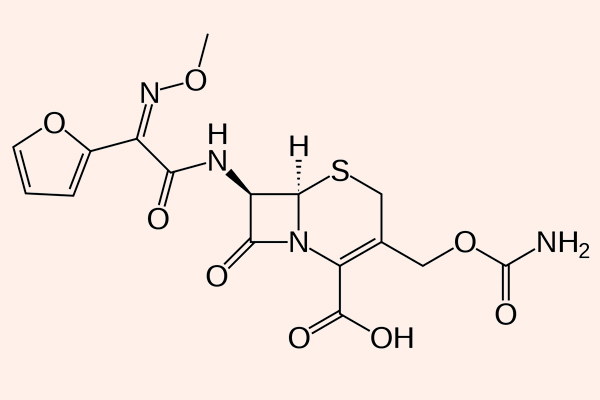
Cephalosporin
Cephalosporin là kháng sinh nhóm β-lactam được sử dụng nhiều nhất trên lâm sàng (khoảng 50%), với số lượng các kháng sinh trong nhóm cũng là nhiều nhất.
Các Cephalosporin có cấu trúc hóa học gần tương đồng với các Penicillin, nhưng cấu trúc vòng 6 cạnh làm giảm sức căng cả hệ vòng, nên khung Cephalosporin thường bền với β-lactamase hơn các Penicillin.
Tác dụng không mong muốn
Dị ứng: Các Cephalosporin cũng có nguy cơ gây ra các phản ứng dị ứng như các Penicillin. Cơ chế tương tự. Bệnh nhân đã dị ứng với Penicillin cũng cơ nguy cơ nhất định dị ứng chéo với Cephalosporin, đặc biệt là các thuốc có cùng nhóm thế R1 trong công thức cấu tạo và lại là các Cephalosporin thế hệ đầu (thế hệ 1, 2). Với trường hợp không cùng nhóm thế R1 thì nguy cơ dị ứng chéo là thấp hơn rất nhiều (khoảng 1%). Bệnh nhân đã có tiền sử dị ứng với Penicillin dạng phản ứng phản vệ không nên sử dụng các Cephalosporin thế hệ 1 và 2, còn với các Cephalosporin thế hệ 3 và 4 thì cần thận trọng khi sử dụng, có thể test dị ứng trên da trước nếu cần thiết.
Các Cephalosporin chứa nhóm methylthiotetrazole trong cấu trúc phân tử (bao gồm những kháng sinh như Cefoperazone và Cefotetan) có độc tính trên hệ tạo máu và có thể gây rối loạn chảy máu. Bệnh nhân bị rối loạn chảy máu do sử dụng kháng sinh có thể sử dụng vitamin K đường uống. Các thuốc nhóm này cũng có thể gây ra phản ứng giống Disulfiram (còn gọi là phản ứng cai rượu), do đó khi sử dụng các thuốc này phải tránh các đồ uống, thức ăn có chứa cồn, nếu không người sử dụng thuốc sẽ có thể gặp phải các tác dụng không mong muốn rất khó chịu. Nguyên nhân của phản ứng này là do thuốc ức chế quá trình giáng hóa ethanol trong cơ thể, làm tích lũy sản phẩm chuyển hóa trung gian của ethanol là acetaldehyde.
Các Cephalosporin thường được chia ra thành 5 thế hệ, phụ thuộc vào phổ kháng khuẩn của nó.
Cephalosporin thế hệ 1
Các đại diện là Cephalexin, Cefazolin, Cephalothin, Cefadroxil…
Phổ tác dụng chủ yếu nằm trên các cầu khuẩn gram dương, như liên cầu và tụ cầu (trừ tụ cầu kháng Methicillin), trực khuẩn gram âm như E.coli, K.pneumoniae và P.mirabilis, các vi khuẩn kị khí như Peptococci và Peptostreptococci.
Cephalexin và Cefazolin là 2 kháng sinh được sử dụng phổ biến nhất trong phân nhóm này, và chúng cũng là 2 kháng sinh duy nhất trong phân nhóm này còn được sử dụng tại Hoa Kỳ. Cephalexin được dùng đường uống, còn Cefazolin được dùng đường tiêm. Chúng đều được thải trừ chủ yếu qua thận và cần được hiệu chỉnh liều phù hợp khi có suy giảm chức năng thận.
Chỉ định của Cephalexin là điều trị nhiễm khuẩn đường tiết niệu hoặc nhiễm khuẩn da và mô mềm do liên cầu hoặc tụ cầu.
Chỉ định của Cefazolin thường là dự phòng trước phẫu thuật. Thuốc phân bố tốt vào hầu hết các mô, nhưng không qua được hàng rào máu não. Bệnh nhân có dị ứng Penicillin mức độ nhẹ (như ngứa, nổi mẩn, phát ban) vẫn có thể sử dụng được Cefazolin.
Cephalosporin thế hệ 2
Các đại diện của phân nhóm này là Cefaclor, Cefuroxime, Cefprozil, Loracarbef… (trong đó chỉ có Cefaclor, Cefuroxime và Cefprozil là còn được sử dụng tại Hoa Kỳ), cùng Cefoxitin và Cefotetan (2 kháng sinh này có phổ trên vi khuẩn kị khí).
Các kháng sinh trong phân nhóm này rất khác nhau về cả dược lực học, dược động học và độc tính. Phổ tác dụng của chúng giống như các Cephalosporin thế hệ 1, nhưng được mở rộng về phía các vi khuẩn gram âm, bao gồm Klebsiella sp. và H.influenzae. Tuy nhiên, Cefoxitin và Cefotetan lại có hoạt lực kém trên H.influenzae và hoạt lực tốt hơn trên B.fragilis và một số chủng Serratia (vi khuẩn kị khí). Các kháng sinh nhóm này không có hoạt tính trên trực khuẩn mủ xanh hoặc các cầu khuẩn ruột (giống các Cephalosporin thế hệ 1).
Các kháng sinh trong nhóm này có đặc tính dược động học đa dạng về cả thời gian bán thải, liên kết protein huyết tương hay thời gian tác dụng. Nhưng có một điểm chung là chúng đều thải trừ qua thận và cần chỉnh liều trong suy giảm chức năng thận.
Cefuroxime là kháng sinh Cephalosporin thế hệ 2 đường uống được sử dụng phổ biến nhất. Các kháng sinh phân nhóm này được chỉ định cho các nhiễm trùng hô hấp cả trên và dưới (đặc biệt là Cefuroxime). Cefoxitin và Cefotetan có thể được sử dụng để điều trị các trường hợp nhiễm trùng hỗn hợp thường có sự tham gia của vi khuẩn kị khí như viêm phúc mạc, viêm túi thừa và viêm vùng chậu. Cefuroxime là kháng sinh Cephalosporin thế hệ 2 duy nhất đạt được nồng độ điều trị trong dịch não tủy, nhưng hiệu quả của nó còn kém xa so với Ceftriaxone và Cefotaxime trong điều trị viêm màng não do vi khuẩn, vậy nên nó không bao giờ được sử dụng trong viêm màng não.
Cephalosporin thế hệ 3
Các đại diện cho phân nhóm này là Cefoperazone, Ceftazidime, Cefotaxime, Ceftriaxone, Cefixime, Cefpodoxime, Cefdinir, Moxalactam… Cefoperazone và Moxalactam không còn được sử dụng ở Hoa Kỳ nữa.
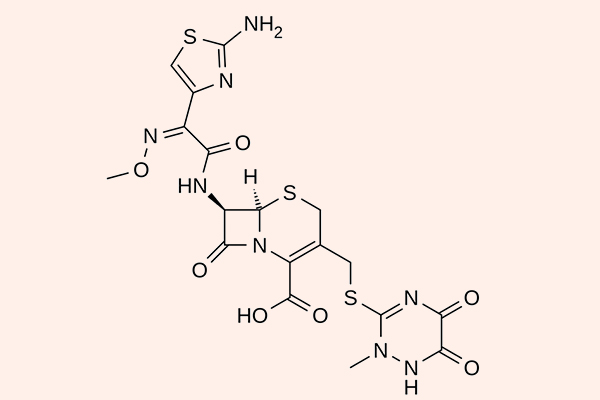
So với thế hệ 2, các kháng sinh thế hệ 3 có phổ kháng khuẩn mở rộng về phía vi khuẩn gram âm, với một số thuốc có thể qua được hàng rào máu não. Tuy nhiên phổ tác dụng trên các vi khuẩn gram dương lại hẹp lại và chúng chỉ còn thể hiện tác dụng tốt nhất là trên phế cầu (2 kháng sinh điều trị nhiễm trùng do phế cầu tốt nhất là Ceftriaxone và Cefotaxime). Phổ tác dụng của các Cephalosporin thế hệ 3 bao gồm các chủng vi khuẩn Citrobacter, S.marcescens, Providencia, Haemophilus và Neisseria. Ceftazidime, Cefoperazone và Moxalactam còn có phổ tác dụng trên trực khuẩn mủ xanh. Các thuốc trong thế hệ 3 này bền vững với β-lactamase hơn thế hệ 2, nhưng chúng vẫn có thể bị thủy phân bởi AmpC β-lactamase hoặc ESBL.
Cefixime, Cefdinir và Cefpodoxime có hoạt tính gần tương tự nhau và là các kháng sinh Cephalosporin thế hệ 3 có thể sử dụng đường uống.
Về dược động học, các thuốc thế hệ 3 này phân bố tốt vào mô và dịch cơ thể, một số thuốc đạt được nồng độ điều trị tốt trong dịch não tủy. Ceftriaxone là thuốc có dược động học thuận lợi với thời gian bán thải dài (7-8 giờ) nên có thể được sử dụng 1 lần mỗi 24 giờ, giúp giảm tần suất dùng thuốc, thuận tiện cho cả bác sĩ và bệnh nhân. Các thuốc còn lại trong nhóm có thời gian bán thải ngắn (t1/2 = 1-1.7 giờ) nên cần được được truyền tĩnh mạch mỗi 6-8 giờ, liều bao nhiêu thì còn tùy thuộc vào mức độ nghiêm trọng của nhiễm trùng. Đa phần các thuốc thế hệ 3 này đều thải trừ chủ yếu qua thận nên cần hiệu chỉnh liều khi suy giảm chức năng thận, trừ 2 trường hợp đặc biệt là Ceftriaxone và Cefoperazone được thải trừ chủ yếu qua mật nên không cần hiệu chỉnh liều ở bệnh nhân suy giảm chức năng thận.
Chỉ định của các Cephalosporin thế hệ 3 là các loại nhiễm trùng nghiêm trọng khi mà nhiều thuốc khác không còn tác dụng. Ceftriaxone và Cefotaxime được phê duyệt cho điều trị viêm màng não do não mô cầu, phế cầu, H.influenzae và các trực khuẩn gram âm đường ruột nhạy cảm, trừ viêm màng não do L.monocytogenes. Ceftriaxone và Cefotaxime có hiệu quả trên các chủng phế cầu kháng Penicillin, nên chúng cũng được chỉ định cho nhiễm trùng đường hô hấp dưới. Ceftriaxone 250 mg tiêm bắp kết hợp với Azithromycin 1 g đường uống 1 liều duy nhất là phác đồ được lựa chọn để điều trị hầu hết các nhiễm trùng lậu cầu (theo khuyến cáo của CDC Hoa Kỳ). Các Cephalosporin thế hệ 3 đường uống (Cefixime, Cefpodoxime và Cefdinir) được sử dụng nhiều trong các nhiễm trùng hô hấp và nhiễm trùng đường tiết niệu ngoài cộng đồng. Các chỉ định khác của các thuốc nhóm này bao gồm điều trị kháng sinh theo kinh nghiệm trong nhiễm trùng huyết (bao gồm cả bệnh nhân suy giảm miễn dịch) và điều trị các nhiễm trùng khác mà Cephalosporin là thuốc an toàn nhất có sẵn.
Cephalosporin thế hệ 4
Các đại diện cho nhóm này là Cefepime và Cefpirome.
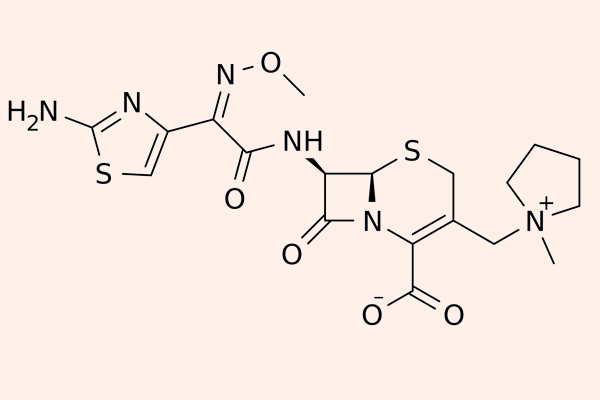
Các thuốc phân nhóm thế hệ 4 này có khả năng đề kháng với β-lactamase cao hơn thế hệ 3 (kể cả các chủng do Enterobacter sản xuất), nhưng chúng vẫn bị thủy phân bởi ESBL. Chúng có hoạt tính tốt trên trực khuẩn mủ xanh, các trực khuẩn đường ruột gram âm, tụ cầu vàng nhạy cảm với Methicillin (MSSA), phế cầu, Haemophilus và Neisseria sp. Thuốc cũng vào được dịch não tủy.
Cefepime được thải trừ chủ yếu qua thận và có dược động học gần tương tự như Ceftazidime. Cần hiệu chỉnh liều thuốc trong suy giảm chức năng thận.
Các thuốc thế hệ này được chỉ định cho các nhiễm trùng nặng và đa kháng, đặc biệt liên quan đến Enterobacter và trực khuẩn mủ xanh. Với trực khuẩn mủ xanh, thường phải tăng liều điều trị hoặc thu hẹp khoảng cách đưa thuốc so với thông thường.
Cephalosporin thế hệ 5
Các Cephalosporin thế hệ 5 còn được gọi là Cephalosporin chống tụ cầu vàng kháng Methicillin (MRSA). Đại diện của phân nhóm này là Ceftaroline, Ceftobiprole và Ceftolozane.
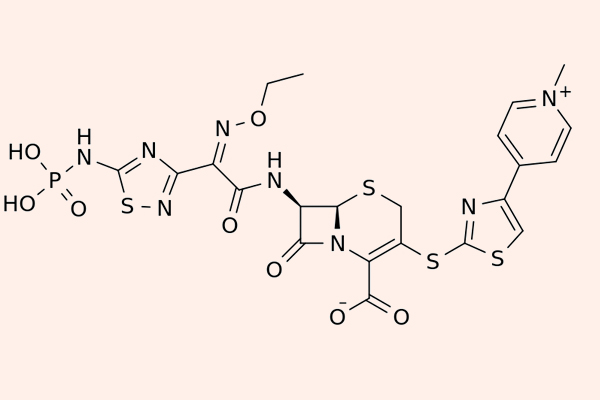
Điểm nổi bật của các kháng sinh phân nhóm này là khả năng liên kết với PBP2a rất tốt, do đó chúng có phổ tác dụng trên MRSA. Tuy nhiên phổ tác dụng trên vi khuẩn gram âm lại kém hơn thế hệ 4 và chúng không được sử dụng để điều trị nhiễm trùng do trực khuẩn mủ xanh. Chúng cũng bị thủy phân bởi ESBL hoặc AmpC β-lactamase.
Ceftaroline hiện được FDA Hoa Kỳ chấp thuận cho điều trị nhiễm trùng da và mô mềm cũng như viêm phổi mắc phải tại cộng đồng. Các nhiễm trùng nặng hơn có thể phối hợp với Aminoside trong điều trị ngoài nhãn (off-label). Ceftaroline thải trừ chủ yếu qua thận nên cần hiệu chỉnh liều ở bệnh nhân suy giảm chức năng thận.
Mặc dù bền vững hơn các Penicillin với các β-lactamase, nhưng tình hình kháng thuốc ngày càng tăng cao đã thúc đẩy các công ty dược phẩm tạo ra các chế phẩm phối hợp giữa Cephalosporin với chất ức chế β-lactamase, chúng được phát triển chủ yếu để chống lại các mầm bệnh gram âm đa kháng. Ceftolozane/Tazobactam và Ceftazidime/Avibactam là 2 ví dụ điển hình cho sự phối hợp này. Chúng đều đã được FDA Hoa Kỳ phê duyệt cho điều trị nhiễm trùng ổ bụng và nhiễm trùng đường tiết niệu có biến chứng. Chúng có hoạt tính mạnh trên trực khuẩn mủ xanh và các trực khuẩn gram âm đường ruột có khả năng sản xuất ESBL và AmpC β-lactamase. Tuy nhiên, chúng vẫn phải “chịu thua” trước “sức mạnh” của metallo-β-lactamase. Với nhiễm trùng ổ bụng có biến chứng, vì thường có sự xuất hiện của vi khuẩn kị khí, nên phối hợp thêm với 1 kháng sinh chống kị khí như Metronidazole. Chúng đều có thời gian bán hủy ngắn (2-3 giờ) và cần phải dùng thuốc mỗi 8 giờ. Cả 2 đều thải trừ chủ yếu qua thận và cần hiệu chỉnh liều ở bệnh nhân suy giảm chức năng thận.
Carbapenem
Carbapenem là nhóm kháng sinh có phổ cực rộng, rộng nhất trong tất cả các β-lactam. Các đại diện của nhóm này là Imipenem/Cilastatin, Meropenem, Doripenem và Ertapenem.
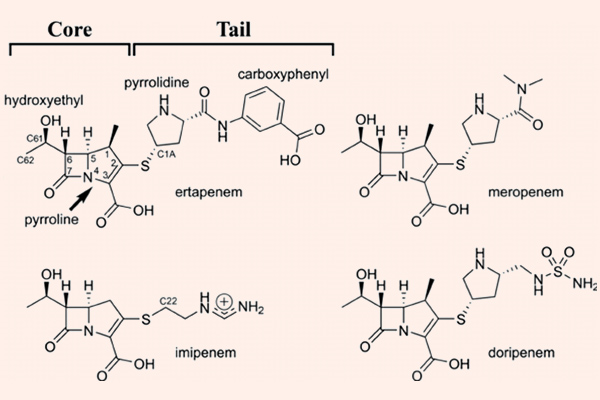
Imipenem là kháng sinh đầu tiên trong nhóm này, có phổ tác dụng tốt trên hầu hết trực khuẩn gram âm (bao gồm cả trực khuẩn mủ xanh), vi khuẩn gram dương, và các vi khuẩn kị khí. Nó bền vững với hầu hết các β-lactamase, ngoại trừ carbapenemase và metallo-β-lactamase. Các Carbapenem không có hiệu quả trên Enterococcus faecium, các chủng tụ cầu kháng Methicillin và C.difficile. Imipenem là một trường hợp khá đặc biệt, nó bị bất hoạt bởi dehydropeptidase ở ống thận, vì thế trên lâm sàng, nó được sử dụng phối hợp với Cilastatin, đây là một chất ức chế dehydropeptidase thận, giúp làm giảm phân hủy Imipenem. Doripenem và Meropenem có phổ tác dụng tương tự như Imipenem, nhưng chúng không bị phá hủy bởi dehydropeptidase thận, và do đó không cần dùng cùng chất ức chế dehydropeptidase thận như Imipenem/Cilastatin. Không giống như 3 carbapenem đã nêu ở trên, Ertapenem không có phổ tác dụng trên trực khuẩn mủ xanh và Acinetobacter.
Các Carbapenem đều phải sử dụng đường tiêm, không thể uống. Phân bố tốt vào nhiều mô và dịch cơ thể, bao gồm cả dịch não tủy, trừ Ertapenem. Tất cả đều được thải trừ chủ yếu qua thận và đều phải hiệu chỉnh liều ở bệnh nhân suy giảm chức năng thận. Các Carbapenem có thời gian bán thải ngắn, chủ yếu là 1-2 giờ. Ertapenem có thời gian bán thải dài nhất, lên tới 4 giờ. Do đó Ertapenem có thể sử dụng theo chế độ liều 1 lần/ngày, trong khi các thuốc còn lại phải theo chế độ liều 3-4 lần/ngày.
Các Carbapenem được chỉ định để điều trị các nhiễm khuẩn nặng và đa kháng được gây ra bởi các chủng vi khuẩn nhạy cảm với Carbapenem, ví dụ như trực khuẩn mủ xanh, nhiễm trùng Enterobacter (trừ Ertapenem), hoặc điều trị nhiễm trùng hỗn hợp cả hiếu khí và kị khí. Carbapenem là một lựa chọn điều trị tốt cho các chủng vi khuẩn gram âm sinh ESBL.
Các tác dụng không mong muốn phổ biến nhất của Carbapenem là buồn nôn và nôn, tiêu chảy, phát ban da và phản ứng ở vị trí tiêm truyền. Đặc biệt là Imipenem còn có thể gây co giật. Cần chú ý tiêu chảy bùng phát và kéo dài liên tục có thể là dấu hiệu của viêm đại tràng giả mạc do nhiễm trùng C.difficile.
Tỷ lệ dị ứng chéo giữa 2 nhóm Carbapenem và Penicillin là dưới 1% (rất thấp).
Monobactam
Đại diện duy nhất của nhóm này hiện nay là Aztreonam, hiện nay cũng không được sử dụng tại Việt Nam.
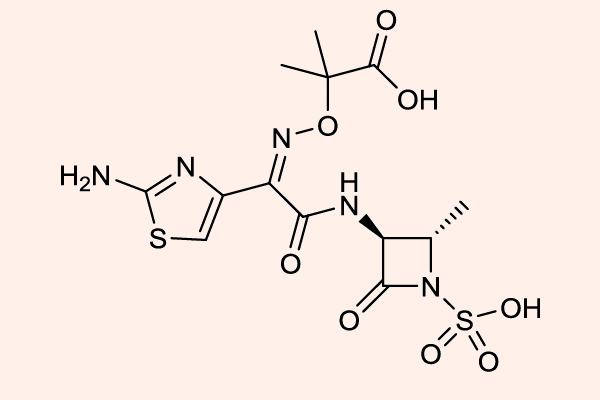
Cấu trúc vòng của Aztreonam rất đặc biệt, nó chỉ chứa duy nhất 1 vòng đơn β-lactam. Aztreonam rất bền với các β-lactamase, trừ AmpC β-lactamase và ESBL. Phổ kháng khuẩn của thuốc này hẹp, giới hạn ở các vi khuẩn hiếu khí gram âm (bao gồm cả trực khuẩn mủ xanh), phổ tác dụng của thuốc khá tương đồng với các Aminoside. Cấu trúc hóa học của Aztreonam gần tương tự với Ceftazidime, và phổ trên vi khuẩn gram âm của nó cũng giống với Cephalosporin thế hệ 3.
Thuốc qua được hàng rào máu não. Aztreonam có thời gian bán thải khoảng 1-2 giờ và kéo dài trong suy giảm chức năng thận. Cần hiệu chỉnh liều ở những bệnh nhân này.
Nguy cơ dị ứng chéo giữa Aztreonam với Penicillin là rất thấp, nhưng nguy cơ dị ứng chéo giữa Aztreonam với Ceftazidime lại cao do cấu trúc hóa học tương tự nhau.
Ở những bệnh nhân bị dị ứng với Penicillin nghiêm trọng (dạng phản ứng phản vệ), Aztreonam vẫn có thể được sử dụng để điều trị các nhiễm trùng nghiêm trọng như nhiễm khuẩn huyết, viêm phổi và viêm màng não gây ra bởi các chủng vi khuẩn hiếu khí gram âm nhạy cảm với Aztreonam.
Các chất ức chế β-lactamase
Các chất ức chế β-lactamase tuy không phải là các kháng sinh, nhưng nó lại rất quan trọng trong thực hành lâm sàng sử dụng các kháng sinh nhóm β-lactam.
Các chất ức chế β-lactamase cổ điển là Acid clavulanic, Sulbactam và Tazobactam. Chúng đều là các chất ức chế β-lactamase mạnh, nhưng không thể ức chế tất cả các β-lactamase. Các chất này có tác dụng bảo vệ kháng sinh β-lactam khỏi sự thủy phân bởi enzyme β-lactamase. Các chất ức chế β-lactamase cổ điển có tác dụng tốt chống lại các β-lactamase Ambler nhóm A (có trong tụ cầu, H.influenzae, lậu cầu, trực khuẩn lỵ, thương hàn, E.coli và K.pneumoniae). Chúng ức chế kém các β-lactamase nhóm C (có trong Enterobacter sp., Citrobacter sp., S.marcescens và trực khuẩn mủ xanh), ngoài ra chúng vẫn có thể ức chế các β-lactamase của B.fragilis và M.catarrhalis.
Chất ức chế β-lactamase không có cấu trúc β-lactam mới là Avibactam có hoạt tính tốt trên β-lactamase Ambler nhóm A, Ambler nhóm B và một số Ambler nhóm D. Biệt dược phối hợp chứa Avibactam là Avycaz (Ceftazidime/Avibactam).
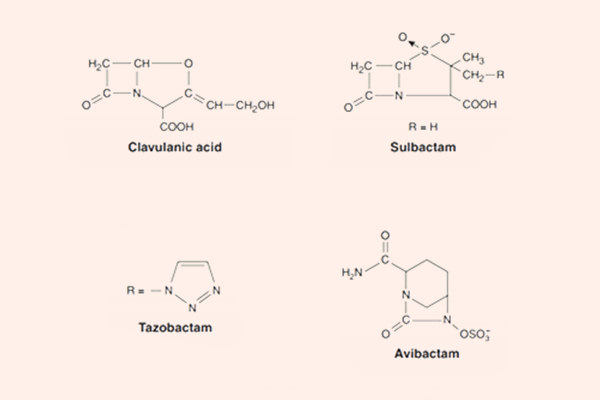
Phối hợp kháng sinh β-lactam với chất ức chế β-lactamase thường được sử dụng trong điều trị kinh nghiệm nhiều loại nhiễm khuẩn, ở cả người bình thường và người suy giảm miễn dịch.
Kháng sinh nhóm Macrolide, Lincosamide và Streptogramin
Tất cả các kháng sinh trong nhóm này tuy thuộc các nhóm khác nhau, nhưng có chung cơ chế tác dụng và cùng một vị trí gắn trên đích tác dụng.
Cơ chế tác dụng
Các kháng sinh trong những nhóm này có tác dụng kìm khuẩn, riêng các Macrolide có thể kìm khuẩn hoặc diệt khuẩn phụ thuộc vào nồng độ cao hay thấp. Tác dụng của các Macrolide được tăng lên trong pH kiềm do tăng tỷ lệ tồn tại dưới dạng phân tử tự do.
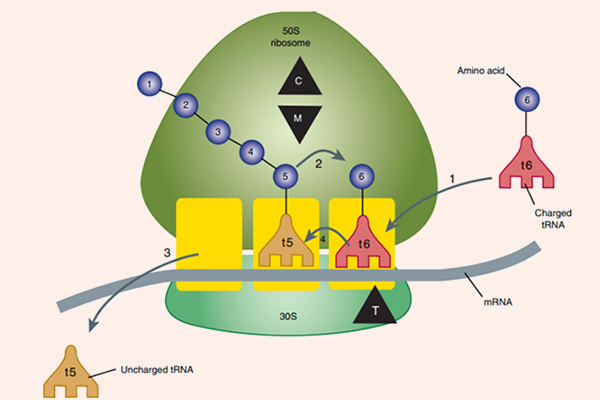
Các thuốc nhóm này đều ức chế tổng hợp protein vi khuẩn thông qua gắn với rARN (ARN ribosome) 50S, từ đó ức chế sự phát triển của vi khuẩn và tạo điều kiện cho hệ miễn dịch tiêu diệt vi khuẩn. Vị trí gắn của phân tử kháng sinh gần với trung tâm peptidyltransferase, và điều này ngăn cản sự kéo dài chuỗi peptide. Kết quả cuối cùng là quá trình tổng hợp protein của vi khuẩn bị ức chế. Ngoài ra, Erythromycin (Macrolide đầu tiên) cũng ức chế sự hình thành tiểu đơn vị ribosome 50S.
Macrolide
Các kháng sinh nhóm này đặc biệt ở chỗ trong cấu trúc có một vòng lactone lớn kết hợp với đường deoxy. Erythromycin là kháng sinh đầu tiên của nhóm này, nó được tìm thấy năm 1952, có nguồn gốc từ xạ khuẩn Streptomyces erythreus, ngày nay nó được đổi tên thành Saccharopolyspora erythraea. Clarithromycin và Azithromycin là các dẫn chất bán tổng hợp từ Erythromycin, với một số đặc tính tốt hơn.
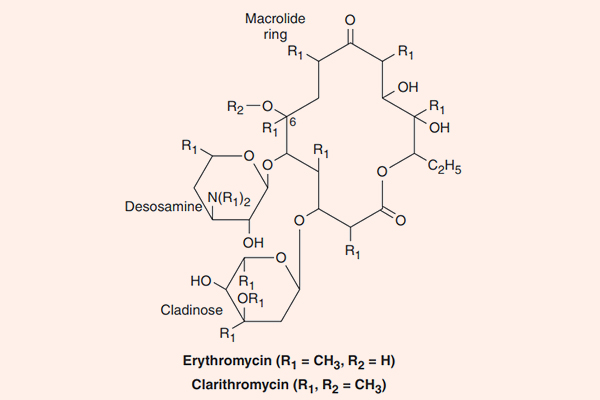
Erythromycin
Phổ tác dụng của Erythromycin bao gồm các chủng vi khuẩn gram dương (phế cầu, tụ cầu, liên cầu và vi khuẩn bạch hầu), vi khuẩn không điển hình (Mycoplasma pneumoniae, L.pneumophila, Chlamydia trachomatis, Chlamydophila psittaci, Chlamydophila pneumoniae, Rickettsiae và L.monocytogenes). Phổ tác dụng trên vi khuẩn gram âm hạn chế, bao gồm xoắn khuẩn giang mai T.pallidum và Campylobacter.
Có 4 cơ chế đề kháng Erythromycin đã được xác định cho đến nay (chỉ ngoại trừ cơ chế “bypass”): Thay đổi đích tác dụng của kháng sinh trên ribosome (nhờ đột biến cấu trúc trên nhiễm sắc thể làm thay đổi cấu trúc của ribosome hoặc nhờ enzyme methylase methyl hóa ribosome), sản xuất enzyme esterase phá hủy thuốc (cơ chế đề kháng của các Enterobacteriaceae), giảm tính thấm của màng tế bào với kháng sinh và bơm tống kháng sinh ra khỏi tế bào. Các gen quy định tính đề kháng kháng sinh của vi khuẩn nằm trên plasmid là chủ yếu. Bơm tống kháng sinh và enzyme methylase là 2 cơ chế kháng thuốc đóng vai trò chính ở vi khuẩn gram dương. Có sự đề kháng chéo giữa Erythromycin với các Macrolide khác. Riêng enzyme methylase còn gây ra sự kháng chéo giữa các Macrolide với Lincosamide và Streptogramin, vì các kháng sinh này vốn có chung 1 vị trí liên kết trên ribosome.
Erythromycin dạng base phải sử dụng dạng bào chế viên bao tan trong ruột nếu uống vì nó không bền trong acid dạ dày. Thức ăn có ảnh hưởng đến hấp thu thuốc. Dạng bào chế stearate và ethylsuccinate bền hơn với acid dày và có hấp thu tốt hơn (do có cấu trúc ester thân dầu hơn).
Erythromycin phân bố rộng vào các dịch và mô trong cơ thể, trừ hệ thần kinh trung ương và dịch não tủy. Thuốc được thải trừ chủ yếu qua gan, nên không cần hiệu chỉnh liều trong suy giảm chức năng thận. Erythromycin có 1 tính chất khá đặc biệt, đó là nó được vận chuyển đến vị trí nhiễm trùng bởi các bạch cầu đa nhân trung tính và đại thực bào. Điều này làm cho tác dụng kháng khuẩn của nó trở nên hiệu quả hơn.
Erythromycin được chỉ định cho bệnh bạch hầu (hiện nay ít gặp vì đã có vaccin phòng ngừa), nhiễm trùng hô hấp (đặc biệt là nhiễm trùng hô hấp dưới), nhiễm trùng mắt và sinh dục do Chlamydia. Tuy rằng có thể sử dụng Erythromycin trong viêm phổi mắc phải tại cộng đồng, tuy nhiên Azithromycin là thuốc được ưu tiên lựa chọn hơn. Hiện nay tỷ lệ phế cầu và M.pneumoniae đề kháng với các Macrolide, trong đó có Erythromycin, đang tăng cao, đặc biệt là ở Việt Nam, nên việc sử dụng các kháng sinh này trong nhiễm trùng hô hấp đang tỏ ra ngày càng kém hiệu quả. Ở những bệnh nhân bị nhiễm trùng do tụ cầu và liên cầu nhưng có dị ứng Penicillin, Erythromycin có thể là một lựa chọn điều trị thay thế tốt. Tuy vậy, tỷ lệ các chủng liên cầu nhóm A đề kháng với Erythromycin đang ngày càng tăng cao.
Các tác dụng không mong muốn phổ biến là trên hệ tiêu hóa như chán ăn, buồn nôn, nôn và tiêu chảy (cần chú ý viêm đại tràng giả mạc do C.difficile). Erythromycin kích thích nhu động dạ dày, do đó nó còn được sử dụng ngoài nhãn (off-label) cho liệt nhẹ dạ dày.
Dạng bào chế có công thức Erythromycin estolate có thể gây viêm gan ứ mật cấp tính (sốt, vàng da, suy gan chức năng).
Erythromycin là thuốc ức chế CYP450 3A4 của gan mạnh và có thể làm tăng nồng độ một số thuốc trong huyết tương nếu các thuốc này được chuyển hóa chủ yếu qua CYP3A4, cần đặc biệt chú ý tới các thuốc có khoảng điều trị hẹp, độc tính cao như Warfarin (gây chảy máu), Theophylin (gây co giật), Cyclosporin (gây nôn, mệt mỏi, vàng da, nhịp tim nhanh, tổn thương thận…) và các statin điều trị rối loạn lipid máu (tăng nguy cơ tiêu cơ vân cấp và suy thận cấp). Erythromycin cũng làm tăng sinh khả dụng của Digoxin nên rất cần chú ý do thuốc này cũng có độc tính cao.
Clarithromycin
Clarithromycin là dẫn chất của Erythromycin bằng bổ sung thêm một nhóm methyl (-CH3) (xem công thức hóa học của Erythromycin và Clarithromycin ở hình trên) và điều này làm tăng độ bền trong acid dịch vị cũng như sinh khả dụng đường uống của thuốc.
Phổ kháng khuẩn của Clarithromycin cũng tương tự như Erythromycin, ngoại trừ việc Clarithromycin tác động tốt trên phức hợp Mycobacterium avium, M.leprae, Toxoplasma gondii và H.influenzae. Tụ cầu và liên cầu kháng Erythromycin cũng kháng chéo với Clarithromycin. Đặc biệt Clarithromycin có phổ tác dụng trên H.pylori nên được sử dụng trong viêm loét dạ dày – tá tràng do vi khuẩn này.
Thời gian bán thải của Clarithromycin dài hơn Erythromycin nên có thể chỉ cần dùng thuốc này 2 lần/ngày. Dạng thuốc giải phóng kéo dài còn cần ít lần dùng hơn, chỉ cần 1 lần/ngày. Clarithromycin được phân bố rộng vào hầu hết các mô, với nồng độ trong mô tương đương hoặc cao hơn nồng độ trong huyết thanh. Thuốc này được chuyển hóa ở gan và thải trừ chủ yếu qua thận (đây là điểm khác biệt với Erythromycin và Azithromycin). Chất chuyển hóa chính của Clarithromycin là 14-hydroxyclarithromycin cũng có hoạt tính kháng khuẩn và thải trừ qua thận. Cần hiệu chỉnh liều ở những bệnh nhân suy giảm chức năng thận có thanh thải creatinine (CrCl) < 30 mL/phút.
Clarithromycin có thể được chỉ định trong nhiễm trùng hô hấp dưới tương tự như Erythromycin, viêm loét dạ dày – tá tràng do H.pylori, nhiễm trùng đường tiết niệu do thải trừ chủ yếu qua nước tiểu.
Clarithromycin cũng là 1 thuốc ức chế CYP3A4 của gan mạnh và có tương tác thuốc tương tự như Erythromycin.
Azithromycin
Azithromycin là một Macrolide mà vòng lactone có 15 nguyên tử (khác với vòng lactone của Erythromycin và Clarithromycin có 14 nguyên tử), là dẫn chất bán tổng hợp từ Erythromycin.
Phổ tác dụng và sử dụng trên lâm sàng của Azithromycin và Clarithromycin là tương tự nhau. Azithromycin có hoạt tính hơi yếu hơn Erythromycin và Clarithromycin trên tụ cầu và liên cầu nhưng hơi mạnh hơn trên H.influenzae. Ngoài ra Azithromycin không có tác dụng tốt trên H.pylori như Clarithromycin. Thuốc vẫn duy trì được tác dụng tốt trên vi khuẩn không điển hình, đặc biệt là Chlamydia.
Sự khác biệt lớn nhất giữa Azithromycin với 2 Macrolide trên là đặc tính dược động học. Nồng độ thuốc khá thấp trong huyết thanh, nhưng rất cao ở trong mô (trừ dịch não tủy) và đại thực bào. Nồng độ thuốc trong mô cao hơn trong huyết thanh từ 10-100 lần, do vậy Azithromycin còn được gọi là kháng sinh có tính “hướng mô”. Thời gian bán thải rất dài (2-4 ngày). Những tính chất độc đáo này cho phép dùng thuốc 1 lần/ngày và rút ngắn thời gian điều trị trong nhiều trường hợp. Ví dụ: 1 liều duy nhất 1 g Azithromycin hiệu quả tương đương 1 liệu trình 7 ngày Doxycycline trong điều trị viêm cổ tử cung và viêm niệu đạo do Chlamydia. Sử dụng Azithromycin với liều nạp 500 mg trong ngày đầu, sau đó là 1 liều 250 mg/ngày trong 4 ngày tiếp theo, hoặc chế độ liều 500 mg/ngày x 3 ngày thường được sử dụng đơn độc hoặc kết hợp với kháng sinh β-lactam để điều trị viêm phổi mắc phải tại cộng đồng.
Azithromycin được hấp thu nhanh và dung nạp tốt qua đường tiêu hóa. Vì chứa vòng lactone có 15 nguyên tử nên Azithromycin không được chuyển hóa qua CYP450 của gan, do đó nó không có tương tác thuốc như Erythromycin và Clarithromycin. Đây là ưu điểm vượt trội của Azithromycin so với 2 kháng sinh đã nêu trên.
Các kháng sinh Macrolide đều làm kéo dài khoảng QT trên điện tâm đồ (ECG) do tác dụng trên kênh kali ở cơ tim, điều này có thể dẫn đến các hậu quả như xoắn đỉnh, rung thất và ngừng tim. Thận trọng khi phối hợp các thuốc này với các thuốc có thể gây kéo dài khoảng QT như thuốc chống loạn nhịp tim nhóm IA (Quinidine…), nhóm III (Amiodarone…), Hydroxychloroquine…
Fidaxomicin
Fidaxomicin là một Macrolide được hấp thu kém qua đường tiêu hóa và được sử dụng trong điều trị viêm đại tràng giả mạc do C.difficile (cùng với 2 lựa chọn điều trị khác là Metronidazole và Vancomycin).
Ketolide
Ketolide là các Macrolide bán tổng hợp có vòng lactone chứa 14 nguyên tử. Phần cấu trúc đường trung tính L-cladinose của Erythromycin được thay thế bằng nhóm 3-keto.
Telithromycin có phổ tác dụng bao gồm S.pyogenes (liên cầu tan huyết β nhóm A), phế cầu, tụ cầu vàng, H.influenzae, M.catarrhalis, Mycoplasma sp., L.pneumophila, Chlamydia sp., H.pylori, lậu cầu, B.fragilis, T.gondii, và một số chủng vi khuẩn lao. Telithromycin không phải cơ chất của bơm tống kháng sinh của vi khuẩn, nên nó lấy lại được tác dụng trên một số chủng vi khuẩn đã kháng các Macrolide thông thường. Ngoài ra cấu trúc đặc biệt cũng cho phép Telithromycin liên kết mạnh hơn với tiêu đơn vị ribosome 50S của một số chủng vi khuẩn.
Về hấp thu, Telithromycin có sinh khả dụng theo đường uống là 57%. Khả năng phân bố vào mô khá tốt. Thuốc được chuyển hóa ở gan và thải trừ qua cả mật và nước tiểu. Telithromycin cũng ức chế CYP3A4 của gan như Erythromycin và Clarithromycin. Nó cũng gây kéo dài khoảng QT trên ECG nhẹ. Telithromycin chỉ được chỉ định duy nhất cho viêm phổi mắc phải tại cộng đồng. Các chỉ định khác của Telithromycin đã bị rút do độc tính trên gan của nó. Telithromycin có thể làm trầm trọng thêm tình trạng yếu cơ nên bị chống chỉ định ở bệnh nhân nhược cơ.
Solithromycin là một ketolide có nhóm thế chứa nguyên tử fluor, còn gọi là fluoroketolide. Thuốc cũng được chỉ định cho điều trị viêm phổi mắc phải tại cộng đồng do kết quả từ 1 thử nghiệm lâm sàng pha III cho thấy nó không thua kém Moxifloxacin trong điều trị bệnh này. Phổ tác dụng của Solithromycin gần như tương tự Telithromycin. Nhưng độc tính trên gan của thuốc thấp hơn nhiều so với Telithromycin, nguyên nhân được cho là do nó thiếu phần cấu trúc pyridine-imidazole.
Lincosamide
Đại diện cho nhóm này là 2 kháng sinh Lincomycin và Clindamycin, trong đó Lincomycin là kháng sinh tự nhiên có nguồn gốc từ Streptomyces lincolnensis, còn Clindamycin là kháng sinh bán tổng hợp từ Lincomycin. Hiện nay, ở nhóm này, người ta chủ yếu dùng Clindamycin.
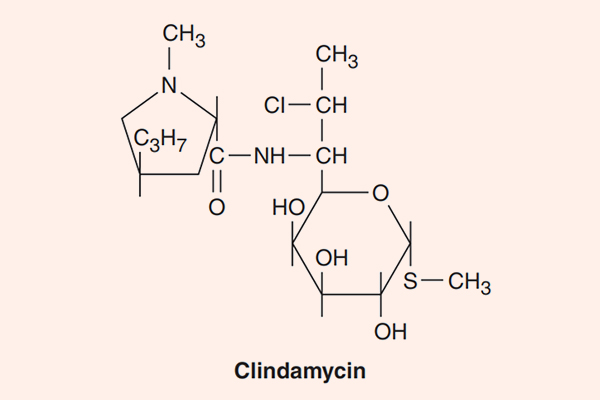
Phổ tác dụng của các kháng sinh nhóm này ưu thế trên vi khuẩn gram dương, bao gồm liên cầu, tụ cầu và phế cầu. Một số chủng MRSA từ cộng đồng còn nhạy cảm với Clindamycin (nhưng các chủng MRSA từ bệnh viện thì không). Clindamycin cũng hiệu quả ở vi khuẩn kị khí như Bacteroides sp., nhưng sự đề kháng đang ngày càng tăng lên, đặc biệt ở các vi khuẩn kị khí gram dương như Clostridium. Vi khuẩn đề kháng với các kháng sinh nhóm này theo cơ chế thay đổi đích tác dụng trên tiểu đơn vị ribosome 50S thường có đề kháng chéo với Macrolide và Streptogramin do chúng có chung vị trí gắn. Một cơ chế đề kháng khác với các kháng sinh nhóm này là sinh enzyme bất hoạt kháng sinh. Các vi khuẩn gram âm hiếu khí đề kháng tự nhiên với thuốc do tính thấm của màng tế bào vi khuẩn với kháng sinh kém
Về phân bố, các thuốc trong nhóm liên kết với protein huyết tương khá cao, thâm nhập tốt vào hầu hết các mô, trừ hệ thần kinh trung ương và dịch não tủy. Các thuốc này cũng phân bố tốt vào các ổ áp xe và được vận chuyển nhờ đại thực bào tương tự như các Macrolide. Về chuyển hóa, chúng được chuyển hóa ở gan, và thải trừ qua cả mật và nước tiểu. Thời gian bán hủy khá ngắn và tăng lên ở bệnh nhân suy thận. Tuy nhiên không cần chỉnh liều cho bệnh nhân suy giảm chức năng thận.
Các thuốc trong nhóm này được chỉ định cho nhiễm trùng da và mô mềm do tụ cầu và liên cầu (đây là chỉ định ưu tiên). Các chỉ định khác là cho các nhiễm trùng do Bacteroides sp. và các vi khuẩn kị khí nhạy cảm khác, thường là điều trị kết hợp với các nhóm kháng sinh khác như Cephalosporin trong các nhiễm trùng hỗn hợp. Các thuốc này cũng được chỉ định cho nhiễm khuẩn xương khớp do chúng thấm vào các vị trí này khá tốt. Clindamycin cũng được chỉ định cho dự phòng viêm nội tâm mạc ở bệnh nhân chuẩn bị làm thủ thuật nha khoa có bệnh van tim nhưng dị ứng với Penicillin ở mức độ đáng kể (thuốc dự phòng bậc 2). Clindamycin + Primaquine được chỉ định là liệu pháp điều trị bậc 2 cho viêm phổi do Pneumocystis jiroveci (trước đây là P.carinii) từ trung bình đến nặng ở bệnh nhân AIDS (Hội chứng suy giảm miễn dịch mắc phải) (đầu tay là Trimethoprim + Sulfamethoxazole). Chỉ định khác cũng ở bệnh nhân AIDS là bệnh não do Toxoplasma (phối hợp với Pyrimethamine).
Các tác dụng không mong muốn phổ biến nhất là tiêu chảy, buồn nôn và phát ban trên da. Sử dụng Clindamycin dễ dẫn đến viêm đại tràng giả mạc do C.difficile nhiều hơn các kháng sinh khác.
Streptogramin
2 kháng sinh hiện đang được sử dụng trong nhóm này không được sử dụng đơn độc mà được kết hợp với nhau, đó là Quinupristin, một Streptogramin B, và Dalfopristin, một Streptogramin A. 2 thuốc này được phối hợp với nhau theo tỷ lệ Quinupristin/Dalfopristin = 3/7.
Phổ tác dụng của chúng bao gồm các cầu khuẩn gram dương, đó là liên cầu (kể cả các chủng kháng đa thuốc), phế cầu (kể cả các chủng kháng Penicillin), tụ cầu (các chủng nhạy cảm hoặc kháng Methicillin) và E.faecium (không phải là E.faecalis vì vi khuẩn này đề kháng tự nhiên với thuốc, có thể là do cơ chế bơm tống thuốc). Các cơ chế đề kháng thuốc đã được phát hiện là thay đổi đích tác dụng của kháng sinh (có thể nhờ enzyme methylase methyl hóa ribosome), sản xuất enzyme để bất hoạt Dalfopristin, hoặc tăng cường số lượng hoặc hoạt động của các bơm tống thuốc.
Về dược động học, Quinupristin và Dalfopristin có nửa đời thải trừ rất ngắn, tương ứng lần lượt là 0.85 và 0.7 giờ. Thải trừ chủ yếu qua mật. Do đó, không cần hiệu chỉnh liều ở bệnh nhân có suy giảm chức năng thận, nhưng ở bệnh nhân có suy giảm chức năng gan thì cần. Chúng ức chế CYP3A4 ở gan đáng kể, dẫn đến có những tương tác thuốc tương tự như Erythromycin và Clarithromycin cần chú ý.
Phối hợp này được chỉ định cho điều trị nhiễm trùng do tụ cầu hoặc các chủng E.faecium kháng Vancomycin.
Tác dụng không mong muốn: Phản ứng tại vị trí truyền tĩnh mạch, hội chứng đau khớp – đau cơ.
Hiện nay, Quinupristin/Dalfopristin không được sử dụng phổ biến do chúng ta có nhiều thuốc khác tốt hơn.
Kháng sinh nhóm Phenicol
Nhóm kháng sinh này gồm 2 thuốc: Chloramphenicol và Thiamphenicol.
Cơ chế tác dụng của các Phenicol là ức chế tổng hợp protein vi khuẩn và có tác dụng kìm khuẩn đối với hầu hết các vi sinh vật nhạy cảm. Cụ thể, nó liên kết thuận nghịch với tiểu đơn vị 50S của ribosome vi khuẩn và ức chế hình thành liên kết peptide (vị trí gắn khác với các Macrolide). Điều này ức chế sự phát triển của vi khuẩn và tạo điều kiện cho hệ miễn dịch tiêu diệt vi khuẩn.
Phổ tác dụng của Chloramphenicol rất rộng, bao gồm nhiều vi khuẩn gram dương và gram âm, cả hiếu khí lẫn kị khí, và có cả vi khuẩn không điển hình (trừ Chlamydia). Tuy nhiên, hiện nay kháng sinh này đã bị đề kháng nhiều.
Các cơ chế đề kháng kháng sinh đã được phát hiện cho đến nay là thay đổi đích tác dụng của kháng sinh (tiểu đơn vị ribosome 50S), sinh enzyme bất hoạt kháng sinh (Chloramphenicol acetyltransferase) và giảm tính thấm của màng tế bào vi khuẩn với kháng sinh. Trong số 3 cơ chế này, cơ chế sinh enzyme bất hoạt kháng sinh là cơ chế quan trọng nhất và có ý nghĩa trên lâm sàng nhất của Chloramphenicol. Các gen quy định tổng hợp enzyme bất hoạt Chloramphenicol được mã hóa trên plasmid và nó có thể được truyền lại cho các vi khuẩn khác thông qua quá trình tiếp hợp.
Trước đây Chloramphenicol được dùng đường toàn thân, nhưng hiện tại do độc tính (gây thiếu máu bất sản và hội chứng xám ở trẻ sơ sinh [gray baby syndrome]), sự đề kháng kháng sinh và có các kháng sinh khác tốt hơn, kháng sinh này hiện nay chỉ còn được sử dụng trên mắt, da, niêm mạc cho các nhiễm khuẩn tại chỗ.
Thiamphenicol tuy ít độc hơn Chloramphenicol, nhưng vẫn không được sử dụng phổ biến.
Kháng sinh nhóm Oxazolidinone
Đây là một nhóm kháng sinh khá mới, tổng hợp hóa học hoàn toàn với 2 đại diện là Linezolid (được sử dụng từ năm 2000) và Tedizolid (được sử dụng từ năm 2014).
Các kháng sinh nhóm này có phổ tác dụng trên các vi khuẩn gram dương là chủ yếu, bao gồm các tụ cầu (bao gồm cả MRSA), liên cầu, cầu khuẩn ruột, trực khuẩn gram dương và một số cầu khuẩn kị khí. Chúng có tác dụng kìm khuẩn, nhưng với liên cầu lại là diệt khuẩn. Ngoài ra, Linezolid còn khá đặc biệt khi có tác dụng trên trực khuẩn lao M.tuberculosis và nó là thuốc bậc 2 trong điều trị lao kháng đa thuốc.
Về cơ chế tác dụng, các kháng sinh nhóm này ức chế tổng hợp protein của vi khuẩn thông qua ức chế tiểu đơn vị ribosome 50S. Chúng ngăn chặn sự hình thành phức hợp ribosome khởi đầu. Vị trí liên kết của kháng sinh với tiểu đơn vị ribosome 50S là rARN 23S, đây là vị trí độc nhất so với các kháng sinh khác. Vậy nên, không có sự đề kháng chéo giữa Linezolid với các nhóm kháng sinh khác. Cơ chế đề kháng với Linezolid được biết duy nhất cho đến nay là do đột biến gen quy định tổng hợp rARN 23S làm giảm ái lực gắn của kháng sinh với ribosome.
Dược động học: Sinh khả dụng theo đường uống của Linezolid là 100% (tương đương dùng đường tĩnh mạch). Thuốc có vào được dịch não tủy 60-70% liều. Linezolid được chuyển hóa theo con đường oxy hóa ở gan, tạo thành 2 chất chuyển hóa không hoạt động. Thời gian bán thải 4-6 giờ.
Linezolid được chỉ định trong điều trị nhiễm trùng do E.faecium kháng Vancomycin, viêm phổi liên quan đến chăm sóc y tế, viêm phổi mắc phải tại cộng đồng, nhiễm trùng da và mô mềm có hoặc không có biến chứng. Linezolid được sử dụng ngoài nhãn (off-label) cho lao đa kháng thuốc (thuốc bậc 2) và nhiễm trùng Nocardia.
Tác dụng không mong muốn nổi bật của Linezolid là ức chế tủy xương. Thường gặp nhất là giảm tiểu cầu, sau đó đến giảm hồng cầu (thiếu máu) và giảm bạch cầu. Tác dụng không mong muốn này dễ xảy ra hơn khi dùng Linezolid trên 2 tuần, nhưng chúng có thể hồi phục sau khi ngừng thuốc. Cũng đã có những báo cáo khi sử dụng phối hợp Linezolid với các thuốc thuốc chống trầm cảm nhóm ức chế tái thu hồi serotonin chọn lọc (SSRIs) gây ra hội chứng serotonin (cảnh báo của FDA).
Tedizolid là kháng sinh cùng nhóm với Linezolid, có cơ chế tác dụng và phổ tác dụng tương tự. Nhưng hiệu lực của thuốc này trên tụ cầu tốt hơn Linezolid. Tỷ lệ liên kết với protein huyết tương của Tedizolid cao hơn Linezolid (70-90% so với 31%).Thời gian bán thải của Tedizolid cũng dài hơn Linezolid. Tedizolid được bào chế dưới dạng dược dụng Tedizolid phosphate.
Tedizolid được chỉ định cho điều trị nhiễm trùng da và mô mềm. Thuốc chưa được chỉ định cho viêm phổi. Tác dụng không mong muốn gây ức chế tủy xương giảm và nguy cơ gây ra hội chứng serotonin khi phối hợp với các thuốc SSRIs của Tedizolid thấp hơn Linezolid, nhưng còn cần thêm bằng chứng lâm sàng cho điều này.
Kháng sinh nhóm Aminoside
Các kháng sinh trong nhóm này bao gồm Streptomycin, Neomycin, Sisomicin, Netilmicin, Kanamycin, Gentamicin, Tobramycin, Amikacin và một số kháng sinh khác. Các thuốc trong nhóm này thường được sử dụng phối hợp với các kháng sinh khác trong điều trị nhiễm khuẩn đa kháng. Trường hợp thường gặp nhất là phối hợp với một β-lactam hoặc kháng sinh ức chế tổng hợp vách tế bào khác như Vancomycin để điều trị nhiễm khuẩn nặng và đa kháng do chúng có hiệp đồng tác dụng, hoặc cũng có thể là phối hợp trong điều trị lao (Streptomycin, Kanamycin, Amikacin).
Các Aminoside là nhóm kháng sinh thân nước, trong phân tử có nhiều phần đường. Phần cấu trúc hexose trong Aminoside có thể là streptidine (trong Streptomycin) hoặc 2-deoxystreptamine (trong các Aminoside khác). Liên kết giữa các phần đường với nhau là liên kết glycoside. Các kháng sinh nhóm này có hoạt tính ở pH kiềm tốt hơn pH acid do khi này chúng tồn tại dưới dạng phân tử là chủ yếu.
Cơ chế tác dụng của các Aminoside là ức chế tiểu đơn vị ribosome 30S của vi khuẩn không hồi phục, từ đó ức chế tổng hợp protein của vi khuẩn và tạo ra tác dụng diệt khuẩn. Cơ chế cụ thể giải thích cho tác dụng diệt khuẩn của Aminoside chưa được tìm ra một cách đầy đủ. Để có thể ức chế được tiểu đơn vị ribosome 30S của vi khuẩn, đầu tiên chúng cần phải vào được trong tế bào. Chúng được khuếch tán theo cơ chế thụ động qua các kênh porin màng ngoài tế bào. Sau đó, chúng được vận chuyển tích cực vào trong tế bào vi khuẩn theo một quá trình phụ thuộc oxy (đây cũng là nguyên nhân Aminoside không có tác dụng trên vi khuẩn kị khí). Năng lượng cho sự vận chuyển thuốc là sự chênh lệch giữa gradient điện hóa 2 bên màng tế bào, cùng với sự tham gia của bơm proton. Nếu trong điều kiện thiếu oxy (yếm khí), thì khả năng vận chuyển thuốc giảm (do quá trình vận chuyển thuốc phụ thuộc oxy). Còn trong điều kiện pH acid, khả năng vận chuyển thuốc cũng giảm do phân tử kháng sinh Aminoside tồn tại dưới dạng ion hóa (do có chứa các nhóm amino mang tính base) là chủ yếu, chứ không phải dạng phân tử. Các kháng sinh ức chế tổng hợp thành tế bào như các β-lactam hoặc Vancomycin có thể giúp tăng cường vận chuyển các Aminoside vào trong tế bào vi khuẩn (do làm thành tế bào vi khuẩn bị suy yếu), tạo ra tác dụng hiệp đồng tăng mức.
Quá trình tổng hợp protein bị ức chế do Aminoside theo ít nhất 3 con đường:
- Ức chế hình thành phức hợp ribosome khởi đầu.
- Gây đọc sai mã trên mARN, tạo ra sự lắp ráp các acid amin không chính xác, dẫn đến hình thành các protein mất chức năng.
- Ức chế sự hình thành polysome từ các monosome.
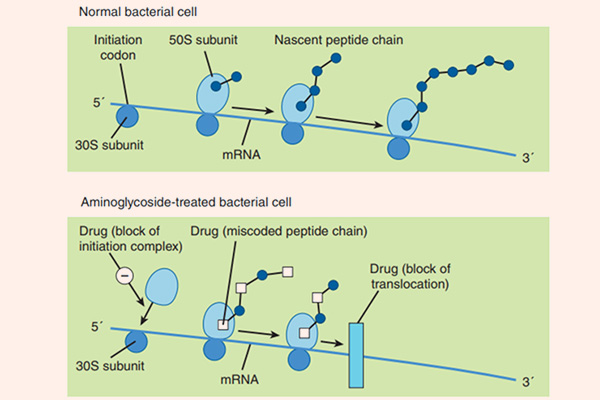
Phổ tác dụng của các kháng sinh nhóm này rơi chủ yếu vào các vi khuẩn gram âm hiếu khí. Một số thuốc có tác dụng tốt trên trực khuẩn mủ xanh là Gentamicin, Tobramycin và Amikacin. Gentamicin, Tobramycin và Amikacin cũng có tác dụng trên một số cầu khuẩn gram dương như tụ cầu và liên cầu, nhưng tác dụng này không mạnh và phải kết hợp với các kháng sinh nhóm khác nếu muốn điều trị nhiễm trùng do các vi khuẩn này. Aminoside không có tác dụng trên vi khuẩn kỵ khí.
Đặc biệt có Streptomycin, Kanamycin và Amikacin có phổ tác dụng trên trực khuẩn lao. Paromomycin có phổ tác dụng trên ký sinh trùng amip. Spectinomycin lại có hoạt tính tốt trên lậu cầu.
Cơ chế đề kháng
Các cơ chế đề kháng với kháng sinh nhóm Aminoside đã được xác định bao gồm:
- Sinh các enzyme bất hoạt kháng sinh: adenyltransferase, acetyltransferase và phosphoryltransferase. Đây là cơ chế đề kháng chính trên lâm sàng. Amikacin là một kháng sinh được bán tổng hợp từ Kanamycin, có khả năng đề kháng với một số cơ chế liên quan đến sinh các enzyme bất hoạt kháng sinh của vi khuẩn tốt hơn nhiều thuốc khác cùng nhóm, do đó nó có tác dụng tốt trên nhiều chủng vi khuẩn đã kháng các Aminoside khác.
- Giảm tính thấm của màng tế bào vi khuẩn với các Aminoside: Đây là kết quả của đột biến hoặc xóa bỏ toàn bộ các kênh porin không cho thuốc đi qua màng ngoài tế bào, hoặc thay đổi gradient điện hóa làm cho quá trình vận chuyển phụ thuộc oxy không hoạt động.
- Đột biến thay đổi vị trí gắn với Aminoside trên tiểu đơn vị ribosome 30S.
Dược động học
Hấp thu: Các Aminoside hấp thu rất kém qua đường uống do kích thước phân tử lớn và quá thân nước, vì vậy trừ trường hợp cần điều trị nhiễm khuẩn đường ruột hoặc chủ động tiêu diệt các vi khuẩn đường ruột thì dùng đường uống, còn các trường hợp khác thì Aminoside đều được dùng đường tiêm tĩnh mạch, tiêm bắp (cho tác dụng toàn thân), hoặc dùng tại chỗ (với tác dụng tại chỗ).
Phân bố: Các Amnioside đều thân nước và phân bố tốt ở các dịch lỏng trong cơ thể, do vậy mà thể tích phân bố thường thấp. Chúng không vào được trong tế bào hay dịch não tủy, nhưng trong viêm màng não, chúng có thể vào được dịch não tủy với nồng độ bằng khoảng 20% nồng độ trong huyết thanh (không cao và sử dụng Aminoside đường toàn thân không có ý nghĩa trong điều trị viêm màng não).
Chuyển hóa và thải trừ: Các kháng sinh này không chuyển hóa mà được thải trừ hoàn toàn qua thận, do đó mức độ thải trừ thuốc có tương quan tỉ lệ thuận với thanh thải creatinine. Cần giảm liều ở bệnh nhân suy giảm chức năng thận.
Ứng dụng dược động học/dược lực học (PK/PD) trong sử dụng thuốc trên lâm sàng
Aminoside là một kháng sinh có chế độ liều rất đặc biệt: Trong đa số các trường hợp, nó sẽ được dùng theo phác đồ 1 lần/ngày. Có 3 lý do chính giải thích cho điều này: Thứ nhất, các Aminoside là kháng sinh diệt khuẩn phụ thuộc nồng độ, có nghĩa là nồng độ càng cao thì diệt khuẩn càng nhiều và càng nhanh, do đó dùng nồng độ tối đa có hiệu quả diệt khuẩn tốt hơn là chia nhỏ liều ra trong ngày. Thứ hai, chúng cũng có tác dụng hậu kháng sinh (PAE) kéo dài, có nghĩa là vi khuẩn vẫn bị ức chế ngay cả khi nồng độ thuốc trong máu đã xuống dưới ngưỡng điều trị. PAE của các Aminoside có thể kéo dài đến vài giờ. Thứ ba, các nghiên cứu lâm sàng cho thấy, với tần suất sử dụng thuốc 1 lần/ngày như vậy, độc tính trên thận của thuốc giảm đi khá nhiều so với chế độ dùng 2-3 lần/ngày.
Tuy nhiên không phải tất cả các trường hợp chúng ta đều sử dụng chế độ liều 1 lần/ngày. Điều này cần có bằng chứng lâm sàng cụ thể trong từng chỉ định của thuốc. Ví dụ: Ở viêm nội tâm mạc do tụ cầu và cầu khuẩn ruột ở bệnh nhân có van nhân tạo, chế độ liều Aminoside 1 lần/ngày không chứng minh được hiệu quả hơn chế độ liều 2-3 lần/ngày. Chế độ liều Aminoside 1 lần/ngày cũng chưa chứng minh được hiệu quả ở phụ nữ có thai, trẻ sơ sinh và người béo phì.
Tác dụng không mong muốn
Các Aminoside đều có độc tính trên thận và thính giác. Độc tính trên thận là có hồi phục, nhưng độc tính trên thính giác là không hồi phục. Độc tính thường xảy ra khi điều trị hơn 5 ngày, liều cao, người cao tuổi, và người suy giảm chức năng thận. Sử dụng phối hợp Aminoside với các thuốc độc thận khác (như lợi tiểu Furosemide, kháng sinh Vancomycin hoặc kháng nấm Amphotericin B) làm tăng độc tính trên thận. Độc tính trên tai có thể là ù tai, mất thính lực, chóng mặt, mất điều hòa và mất thăng bằng. Độc tính của mỗi kháng sinh có mức độ khác nhau. Neomycin, Kanamycin và Amikacin độc với thính giác nhất. Streptomycin và Gentamicin độc với tiền đình nhất. Neomycin, Tobramycin và Gentamicin độc với thận nhất. Tuy nhiên các độc tính của Neomycin không có ý nghĩa vì nó không bao giờ được sử dụng theo đường toàn thân.
Các Aminoside có thể ức chế thần kinh – cơ tương tự các curae, đặc biệt là ở nồng độ cao. Nguy cơ nguy hiểm nhất là ngừng thở do liệt cơ hô hấp. Đây là một tình trạng cực kỳ nguy hiểm và có thể gây tử vong. Cấp cứu tình trạng này bằng Calcium gluconate hoặc Neostigmine.
Quá mẫn ít khi xảy ra.
Chỉ định
Các Aminoside dùng đường toàn thân có thể được chỉ định cho các nhiễm trùng do các chủng vi khuẩn gram âm kháng thuốc. Chúng thường được phối hợp với các kháng sinh nhóm khác, nhất là nhóm kháng sinh ức chế tổng hợp thành tế bào như β-lactam. Ví dụ: Ampicillin + Gentamicin được chỉ định cho điều trị viêm nội tâm mạc do cầu khuẩn ruột và rút ngắn thời gian điều trị trong viêm nội tâm mạc do liên cầu S.viridans. Thường chỉ sử dụng đơn độc các thuốc nhóm này trong nhiễm trùng đường tiết niệu không biến chứng.
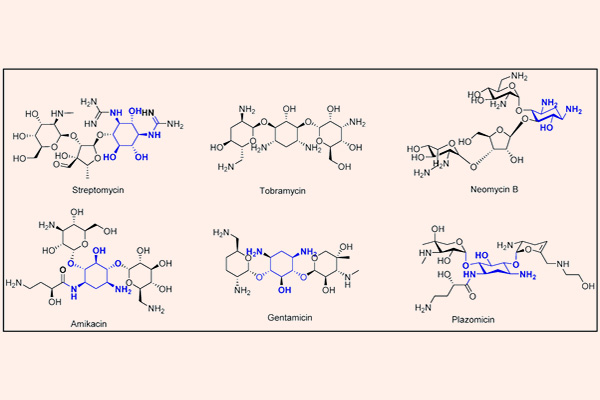
Streptomycin: Đây là kháng sinh đầu tiên được phát hiện trong nhóm này. Nó có nguồn gốc tự nhiên từ chủng Streptomyces griseus, được sử dụng phổ biến như là thuốc đầu tay trong điều trị lao do M.tuberculosis, phối hợp với 4 thuốc khác. Hiện nay do tình trạng kháng thuốc ngày càng tăng, nhiều nước trên thế giới đã xếp Streptomycin vào hàng các thuốc bậc hai. Các chỉ định khác là dịch hạch, sốt thỏ và sốt Malta, nhưng phải sử dụng phối hợp với các Tetracycline đường uống, và các chỉ định này cũng không phổ biến.
Gentamicin: Đây là một kháng sinh được tạo thành từ hỗn hợp 3 chất C1, C1A và C2, chúng có nguồn gốc từ loài Micromonospora purpurea. Chỉ định cho các nhiễm trùng gram âm nặng và kháng thuốc như nhiễm trùng do trực khuẩn mủ xanh, Enterobacter sp., Serratia marcescens, Proteus sp., Acinetobacter sp. và Klebsiella sp. Hầu như luôn phối hợp Gentamicin với kháng sinh nhóm khác, trừ trường hợp nhiễm khuẩn đường tiết niệu. Phối hợp Gentamicin với kháng sinh ức chế tổng hợp vách tế bào như các β-lactam và Vancomycin được chỉ định cho các trường hợp như viêm nội tâm mạc do tụ cầu, liên cầu hoặc cầu khuẩn ruột. Có thể sử dụng kháng sinh này theo đường tại chỗ. Tiêm trực tiếp vào dịch não tủy có thể giúp nồng độ thuốc đạt đến ngưỡng điều trị ở đây, chỉ định cho điều trị viêm màng não.
Tobramycin: Phổ của kháng sinh này rộng hơn Gentamicin. Có thể có sự đề kháng chéo giữa Tobramycin và Gentamicin. Tobramycin có hoạt tính cao hơn Gentamicin trên trực khuẩn mủ xanh và E.faecalis. Chỉ định cho các nhiễm trùng gram âm nặng và đa kháng, đặc biệt do trực khuẩn mủ xanh gây ra. Thuốc có thể dùng theo nhiều đường: tĩnh mạch, tiêm bắp, nhỏ mắt.
Amikacin: Kháng sinh này có nhiều ưu điểm hơn so với các Aminoside khác. Hoạt tính của nó trên trực khuẩn mủ xanh cũng cao hơn Tobramycin. Chỉ định cho các nhiễm trùng gram âm nặng và đa kháng, đặc biệt do trực khuẩn mủ xanh gây ra. Nó cũng là thuốc bậc hai trong điều trị lao, sau khi điều trị đầu tay thất bại.
Neomycin: Không có phổ tác dụng trên trực khuẩn mủ xanh. Kháng sinh này chỉ sử dụng đường uống và tại chỗ do độc tính cao. Đường uống được sử dụng khi cần tiêu diệt sạch hệ khuẩn chí đường ruột (ví dụ: bệnh nhân hôn mê gan), hoặc làm sạch đại tràng trước phẫu thuật đường tiêu hóa.
Kanamycin: Không có phổ tác dụng trên trực khuẩn mủ xanh. Thuốc này cũng được chỉ định cho làm sạch đại tràng trước phẫu thuật đường tiêu hóa. Chỉ định khác là thuốc bậc 2 trong lao đa kháng thuốc. Có đề kháng chéo giữa Kanamycin và Neomycin.
Paromomycin: Chỉ định cho bệnh leishmania nội tạng (dùng đường tĩnh mạch), bệnh lỵ amip ở đại tràng và đôi lúc được sử dụng trong nhiễm trùng đường ruột do ký sinh trùng khác (dùng đường uống).
Plazomicin: Đây là Aminoside mới được phê duyệt bởi FDA năm 2018. Nó là một kháng sinh tổng hợp có nguồn gốc từ Sisomicin, một Aminoside hiện nay không còn được sử dụng nữa. Plazomicin bền vững với hầu hết các enzyme phá hủy Aminoside của vi khuẩn, do đó nó có tác dụng trên nhiều chủng vi khuẩn đã kháng các Aminoside khác. Chỉ định của thuốc là nhiễm trùng đường tiết niệu biến chứng hoặc nhiễm khuẩn huyết do Enterobacteriaceae đa kháng, kể cả chủng đã kháng Carbapenem.
Spectinomycin:
Đây không hẳn là một Aminoside, nhưng có liên quan cấu trúc với Aminoside nên được xếp vào mục này. Spectinomycin là một aminocyclitol, nó thiếu nhóm đường amin và liên kết glycoside.
Phổ tác dụng của thuốc bao phủ nhiều vi khuẩn gram dương và gram âm, nhưng chỉ định chủ yếu của kháng sinh này là bệnh lậu kháng thuốc hoặc lậu ở bệnh nhân dị ứng Penicillin. Không nên sử dụng Spectinomycin cho điều trị nhiễm trùng lậu cầu ở hầu họng do tỷ lệ thất bại cao. Chế độ liều chỉ dùng 1 liều duy nhất.
Kháng sinh nhóm Tetracycline
Các dạng base của Tetracycline rất khó tan trong nước, vậy nên chúng thường được sử dụng dưới dạng muối hydrochloride (HCl) để tăng độ tan và tăng độ ổn định trong dung dịch.
Dược lực học và cơ chế đề kháng
Các Tetracycline là các kháng sinh có phổ tác dụng rộng nhất trong số tất cả các kháng sinh hiện có. Nó có tác dụng trên nhiều vi khuẩn gram âm và gram dương, hiếu khí và kị khí, vi khuẩn không điển hình, và cả một số ký sinh trùng nữa.
Cơ chế tác dụng của các kháng sinh nhóm này là ức chế tổng hợp protein thông qua ức chế thuận nghịch tiểu đơn vị 30S của ribosome vi khuẩn, điều này làm giảm khả năng liên kết của aminoacyl-tARN với vị trí tiếp nhận trên phức hợp mARN-ribosome. Như vậy các acid amin mới không thể được thêm vào chuỗi peptide đang được tổng hợp.
Có 3 cơ chế đề kháng với các Tetracycline đã được nghiên cứu chi tiết, đó là: Thay đổi đích tác dụng của kháng sinh (tiểu đơn vị ribosome 30S), sản xuất enzyme bất hoạt kháng sinh và hình thành các bơm tống kháng sinh. Cơ chế thứ nhất và thứ ba là 2 cơ chế quan trọng nhất trên lâm sàng. Với cơ chế bơm tống thuốc, các Tetracycline thế hệ sau, bao gồm Doxycycline, Minocycline và Tigecycline, thường ít nhạy cảm với bơm tống thuốc của vi khuẩn hơn, và do đó nó thường có tác dụng trên các chủng vi khuẩn đã kháng các Tetracycline cổ điển. Tuy nhiên, một số vi khuẩn có bơm tống đa thuốc, đó là trực khuẩn mủ xanh và Proteus sp., các vi khuẩn này có thể đề kháng với tất cả các Tetracycline. Với cơ chế thay đổi đích tác dụng, Tigecycline thường không bị ảnh hưởng bởi cơ chế này như các Tetracycline khác.
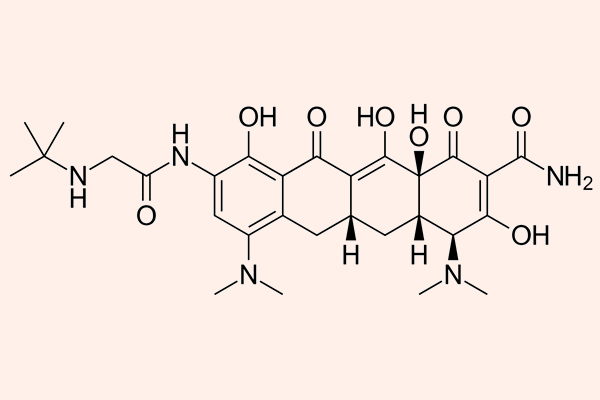
Dược động học
Dược động học các thuốc nhìn chung rất khác nhau.
Hấp thu: Doxycycline và Minocycline là 2 Tetracycline có sinh khả dụng đường uống tốt nhất (95-100%). Các Tetracycline cổ điển có sinh khả dụng đường uống thấp hơn. Tigecycline gần như không hấp thu qua đường uống và phải dùng đường tĩnh mạch. Các kháng sinh nhóm này đều có khả năng tạo phức chelate với các ion kim loại hóa trị II và III (Ca2+, Mg2+, Fe2+, Al3+), điều này làm giảm hấp thu và sinh khả dụng của thuốc. Hấp thu của Doxycycline và Minocycline không bị ảnh hưởng bởi thức ăn, nhưng Tetracycline và Demeclocycline thì có.
Phân bố: Phân bố tốt vào các mô và dịch trong cơ thể, trừ dịch não tủy. Liên kết với protein huyết tương 40-80%. Thuốc qua được hàng rào nhau thai và sữa mẹ. Đặc biệt, các thuốc nhóm này có thể gắn vào Ca trong xương và răng, gây mất thẩm mỹ, ức chế phát triển xương. Đây là nguyên nhân thuốc bị chống chỉ định cho trẻ em dưới 8 tuổi và phụ nữ có thai.
Thải trừ: Các Tetracycline được thải trừ qua cả mật và nước tiểu, nhìn chung thải trừ qua mật chiếm ưu thế hơn một chút. Đó là lý do nhiều thuốc không cần hiệu chỉnh liều trong suy giảm chức năng thận.
Các Tetracycline cổ điển có thời gian tác dụng ngắn (t1/2 = 6-8 giờ), Còn các Tetracycline mới hơn có thời gian tác dụng dài hơn (Doxycycline và Minocycline có t1/2 = 16-18 giờ, Tigecycline có t1/2 = 36 giờ)
Chỉ định
Các kháng sinh nhóm này được chỉ định cho bệnh Lyme, nhiễm trùng do Rickettsia, Borrelia sp., Anaplasma phagocytophilum, Ehrlichia sp., M.pneumoniae, Chlamydia, một số xoắn khuẩn (như giang mai), H.pylori, phẩy khuẩn tả, sốt thỏ, sốt Malta và dịch hạch. Chỉ định khác bao gồm dự phòng nhiễm trùng Plasmodium falciparum (gây bệnh sốt rét ác tính), điều trị mụn trứng cá, đợt cấp của viêm phế quản mạn, viêm phổi mắc phải tại cộng đồng. Tuy có nhiều ứng dụng lâm sàng như vậy, nhưng hiện nay tỷ lệ kháng các Tetracycline, đặc biệt là các Tetracycline cổ điển, đang ở mức rất cao. Nguyên nhân một phần có thể là do việc sử dụng kháng sinh này bừa bãi ở cộng đồng trước đây.
Chỉ định đặc biệt: Demeclocycline được sử dụng ngoài nhãn (off-label) để ức chế hormon chống bài niệu (ADH) ở ống thận.
Tigecycline: Đây là kháng sinh mới nhất trong nhóm này, với phổ tác dụng rộng trên nhiều chủng đã kháng các Tetracycline khác. Đặc biệt nó có tác dụng trên cả các tụ cầu đã kháng Methicillin và Vancomycin, Enterobacteriaceae, các chủng Acinetobacter sp. đa kháng thuốc, các vi khuẩn này có thể gây ra nhiều nhiễm trùng bệnh viện nghiêm trọng.
Chỉ định của thuốc này là nhiễm trùng da và cấu trúc da, nhiễm trùng ổ bụng và viêm phổi mắc phải tại cộng đồng. Viêm phổi mắc phải tại bệnh viện và viêm phổi thở máy chỉ sử dụng Tigecycline khi không thể sử dụng các kháng sinh khác (cảnh báo FDA Hoa Kỳ), do thuốc làm tăng nhẹ nguy cơ tử vong, được quan sát thấy trong các thử nghiệm lâm sàng.
Tác dụng không mong muốn
- Rối loạn tiêu hóa
Buồn nôn, nôn và tiêu chảy là các tác dụng không mong muốn phổ biến nhất. Chú ý nguy cơ viêm đại tràng giả mạc do C.difficile, nhưng nguy cơ của các kháng sinh Tetracycline thấp hơn các nhóm kháng sinh khác.
- Tác dụng phụ trên cấu trúc xương và răng
Thuốc có thể gắn với calcium trong xương và răng, gây đổi màu, mất thẩm mỹ và ức chế phát triển xương. Chống chỉ định cho phụ nữ có thai hoặc trẻ em dưới 8 tuổi.
- Các tác dụng không mong muốn khác
Các tác dụng không mong muốn khác của Tetracycline bao gồm: Suy giảm chức năng gan (đặc biệt với phụ nữ có thai, bệnh nhân có tiền sử bệnh gan và dùng liều cao tĩnh mạch), nhiễm toan ống thận, hội chứng Fanconi, huyết khối tĩnh mạch (với đường dùng tĩnh mạch), nhạy cảm với ánh sáng, tia cực tím (đặc biệt ở người da trắng), ù tai…
Kháng sinh nhóm Peptide
Các kháng sinh trong nhóm này có cấu trúc peptide, tức là có nhiều đơn phân acid amin liên kết với nhau. Các acid amin trong phân tử có thể là đồng phân D hoặc L, nhưng số lượng đồng phần D nhiều hơn. Do là peptide nên thuốc thường có phân tử lượng lớn hơn so với các nhóm kháng sinh khác, đồng thời khó bị chuyển hóa. Trong phân tử thuốc, ngoài phần acid amin còn có thể có thêm phần đường hoặc lipid, hoặc cả hai.
Đây là nhóm kháng sinh thường có hoạt tính mạnh, diệt khuẩn tốt, nhưng đi kèm với đó là độc tính thường cao. Chúng ít khi là các kháng sinh sử dụng đầu tay trong điều trị kháng sinh theo kinh nghiệm.
Các kháng sinh Peptide có thể chia ra thành 4 phân loại nhỏ hơn, mỗi loại lại có cơ chế tác dụng, cách sử dụng trên lâm sàng hoàn toàn khác nhau:
- Glycopeptide: Trong phân tử có gắn các nhóm đường.
- Lipopeptide: Ngoài phần cấu trúc peptide còn có phần cấu trúc thân lipid.
- Cyclopeptide: Có cấu trúc là một peptide vòng.
- Polypeptide thiazolidic: Trong cấu trúc ngoài phần peptide còn có liên kết với một gốc là dẫn xuất của vòng 1,3-thiazol.
Glycopeptide
Các đại diện của nhóm này là Vancomycin, Teicoplanin, Telavancin, Dalbavancin và Oritavancin.
Vancomycin
Vancomycin là một kháng sinh tự nhiên, được phân lập từ loài Amycolatopsis orientalis. Nó chỉ có tác dụng trên các vi khuẩn gram dương do kích thước phân tử lớn và tính thấm kém với màng tế bào vi khuẩn gram âm.
Cơ chế tác dụng chung của Vancomycin cũng như các kháng sinh khác trong nhóm Glycopeptide này là ức chế tổng hợp thành tế bào. Tuy nhiên, cơ chế phân tử của nó khác với các kháng sinh nhóm β-lactam. Các kháng sinh nhóm này ức chế tổng hợp thành tế bào ở giai đoạn sớm hơn so với các β-lactam. Chúng gắn vào đầu D-Ala-D-Ala của chuỗi pentapeptide (xem lại phần viết về kháng sinh nhóm β-lactam) và ức chế sự hình thành liên kết chéo trong chuỗi peptidoglycan.
Một cơ chế đề kháng quan trọng với các kháng sinh nhóm này trên lâm sàng là thay đổi đích tác dụng của kháng sinh, cụ thể là thay đổi cấu trúc đầu D-Ala-D-Ala của chuỗi pentapeptide bằng D-Ala-D-Lac (lactate), làm mất một liên kết hydro trong liên kết so với đầu D-Ala-D-Ala.
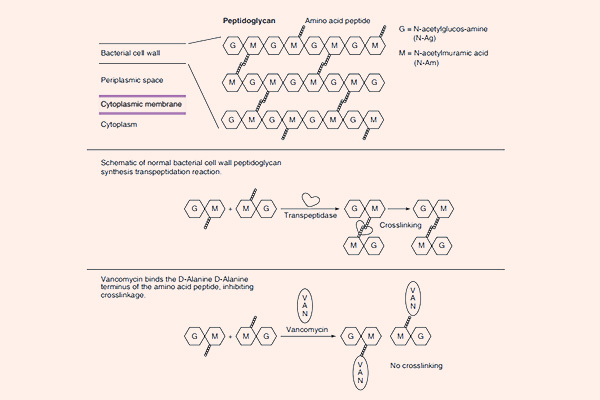
Vancomycin chỉ có phổ trên vi khuẩn gram dương. Tụ cầu nhìn chung nhạy cảm với Vancomycin, kể cả các chủng đã kháng Methicillin, tuy nhiên giá trị MIC (nồng độ ức chế tối thiểu) của tụ cầu vàng kháng Methicillin với Vancomycin đang tăng dần. Đây là kháng sinh diệt khuẩn chậm, phụ thuộc thời gian, chỉ có tác dụng với các vi khuẩn đang phát triển và nhân lên. Các cầu khuẩn ruột gram dương khác cùng nằm trong phổ tác dụng của Vancomycin là E.faecium và E.faecalis. C.difficile và các vi khuẩn kỵ khí gram dương khác cùng nhạy cảm với Vancomycin.
Thuốc được dùng đường tĩnh mạch, trừ trường hợp điều trị viêm đại tràng giả mạc do C.difficile thì dùng đường uống (do thuốc không hấp thu theo đường uống). Phân bố của Vancomycin khá rộng trong cơ thể. Vancomycin qua được hàng rào máu não không nhiều, vào nhiều hơn khi màng não bị viêm. Thải trừ chủ yếu thông qua lọc ở cầu thận. Cần hiệu chỉnh liều ở bệnh nhân suy giảm chức năng thận.
Vancomycin được chỉ định chủ yếu cho nhiễm trùng huyết và viêm nội tâm mạc do tụ cầu kháng Methicillin, viêm nội tâm mạc do cầu khuẩn ruột ở bệnh nhân bị dị ứng Penicillin nghiêm trọng (phản ứng phản vệ) (có phối hợp với Aminoside), viêm đại tràng giả mạc do C.difficile mức độ nặng.
Thuốc có độc tính trên thận và thính giác. Độc tính trên thận tăng lên khi phối hợp thuốc này với các thuốc độc với thận khác như Aminoside.
Một trong những phản ứng phổ biến nhất khi sử dụng Vancomycin truyền tĩnh mạch là hội chứng “người đỏ” (red man). Lý do là bởi truyền Vancomycin quá nhanh, gây giải phóng histamine. Khắc phục tác dụng không mong muốn này bằng kéo dài thời gian truyền và sử dụng thuốc kháng histamine H1 như Diphenhydramine.
Teicoplanin
Kháng sinh này giống hệt Vancomycin về phổ kháng khuẩn và cơ chế tác dụng. Teicoplanin có thể được tiêm bắp hoặc truyền tĩnh mạch. Thời gian bán thải thuốc dài (45-70 giờ), chỉ sử dụng 1 lần/ngày. Thuốc được lưu hành ở châu Âu, nhưng Hoa Kỳ thì không.
Telavancin
Telavancin là một lipoglycopeptide bán tổng hợp từ Vancomycin. Phổ kháng khuẩn của Telavancin giống như Vancomycin, nhưng còn giữ được hoạt tính trên nhiều chủng vi khuẩn đã giảm nhạy cảm với Vancomycin.
Telavancin có 2 cơ chế tác dụng. Cơ chế thứ nhất cũng giống như Vancomycin, Telavancin ức chế tổng hợp thành tế bào bằng cách gắn vào D-Ala-D-Ala. Ngoài ra, cơ chế thứ hai là nó phá vỡ điện thế màng và tăng tính thấm màng tế bào vi khuẩn. Thời gian bán thải là 8 giờ, dùng 1 lần/ngày.
Thuốc được phê duyệt cho nhiễm trùng da và mô mềm có biến chứng và viêm phổi mắc phải tại bệnh viện.
Thuốc cũng có độc tính trên thận như Vancomycin.
Không dùng thuốc cho phụ nữ có thai.
Dalbavancin và Oritavancin
Dalbavancin và Oritavancin là các lipoglycopeptide bán tổng hợp từ Teicoplanin.
Cơ chế tác dụng của 2 thuốc này cũng giống Teicoplanin và Vancomycin, nhưng Oritavancin còn có một cơ chế nữa, đó là làm rối loạn tính thấm màng tế bào và ức chế tổng hợp ARN.
2 thuốc này có hoạt lực trên các vi khuẩn gram dương cao hơn (MIC thấp hơn) và còn giữ được tác dụng tốt trên các chủng tụ cầu kháng Methicillin nhưng nhạy cảm trung gian với Vancomycin. Đối với các chủng cầu khuẩn ruột kháng Vancomycin (Vancomycin Resistant Enterococcus, hay VRE), Dalbavancin không có tác dụng, còn Oritavancin chỉ có tác dụng in vitro, còn hiệu quả lâm sàng không rõ ràng.
Cả 2 loại thuốc này đều có thời gian bán thải rất dài (> 10 ngày), cho phép dùng thuốc 1 lần/tuần.
2 thuốc này được chỉ định cho điều trị nhiễm trùng da và mô mềm. Mỗi thuốc có một chế độ liều đặc trưng riêng.
Không cần hiệu chỉnh liều cả 2 thuốc này ở bệnh nhân suy giảm chức năng gan hoặc thận mức độ từ nhẹ đến trung bình.
Lipopeptide
Các đại diện cho nhóm kháng sinh này là Daptomycin, Polymyxin B và Polymyxin E (Colistin).
Daptomycin
Daptomycin là một lipopeptide vòng có nguồn gốc từ loài Streptomyces roseosporus.
Phổ tác dụng của Daptomycin là giống với Vancomycin, ngoại trừ việc nó có tác dụng tốt trên cả các chủng cầu khuẩn ruột và tụ cầu vàng đã kháng Vancomycin. Nó có hoạt tính diệt khuẩn nhanh hơn Vancomycin in vitro.
Cơ chế tác dụng đầy đủ của Daptomycin chưa thực sự rõ ràng, nhưng có bằng chứng cho thấy rằng Daptomycin được tích hợp vào màng tế bào bằng cách chèn vào màng phần gốc lipid của nó. Quá trình này có sự tham gia của ion calcium. Các phân tử Daptomycin sau khi tích hợp vào màng tế bào sẽ hình thành một cấu trúc giống như một kênh ion, gây rò rỉ ion K+ ra khỏi tế bào vi khuẩn, làm chết tế bào (nhưng không có sự ly giải tế bào).
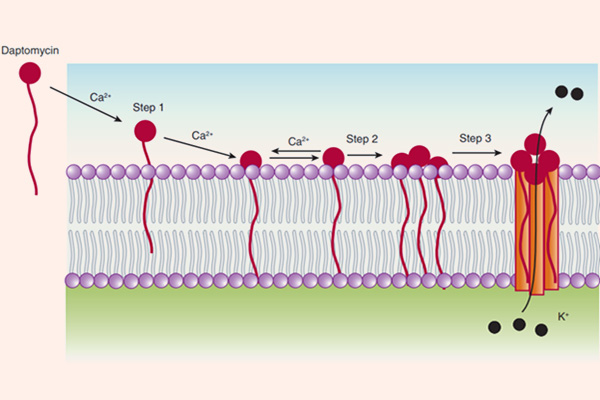
Chỉ định của Daptomycin là nhiễm trùng da và mô mềm, nhiễm trùng huyết, viêm nội tâm mạc và một số nhiễm trùng nghiêm trọng khác. Daptomycin không thể dùng để điều trị viêm phổi, vì nó bị bất hoạt bởi chất diện hoạt trong phổi.
Daptomycin được thải trừ qua thận hoàn toàn và cần hiệu chỉnh liều ở bệnh nhân suy giảm chức năng thận.
Tác dụng không mong muốn nổi bật của thuốc này là gây ra bệnh cơ và tiêu cơ vân, do đó cần theo dõi nồng độ creatine phosphokinase (CPK) thường xuyên.
Các Polymyxin
Các Polymyxin bao gồm Polymyxin B và Polymyxin E (Colistin). Phổ của chúng tập trung chủ yếu trên các vi khuẩn gram âm, trừ các chủng Proteus sp. và Neisseria sp. Các kháng sinh nhóm này cũng ức chế màng tế bào vi khuẩn bằng cách gắn vào màng tế bào và phá vỡ nó. Chúng cũng gắn và làm bất hoạt nội độc tố vi khuẩn.
Các kháng sinh nhóm này có độc tính cao trên thận. Polymyxin B chỉ được dùng tại chỗ, còn Colistin chỉ được xem là giải pháp cuối cùng để điều trị các vi khuẩn gram âm đa kháng, khi không còn lựa chọn nào khác (A.baumannii, trực khuẩn mủ xanh và các Enterobacteriaceae). Polymyxin B thường được phối hợp với Bacitracin hoặc Neomycin, hoặc cả hai.
Cyclopeptide
Ở đây chúng ta nói đến 2 kháng sinh là Tyrocidin và Gramicidin. Chúng là các peptide có cấu tạo vòng, bản thân mỗi kháng sinh lại là hỗn hợp của những loại khác nhau (như Tyrocidin A, B, C, D và E; Gramicidin A, B và C). Đây là các kháng sinh tại chỗ được ưa dùng trong các nhiễm khuẩn ở khu vực miệng và họng.
Các kháng sinh này có phổ tác dụng tốt trên các vi khuẩn gram dương như tụ cầu và liên cầu. Chúng có thể được chỉ định cho các trường hợp viêm họng, viêm thanh quản, viêm lợi… do vi khuẩn.
Dùng thuốc tại chỗ rất an toàn và hiếm khi gặp phải tác dụng không mong muốn nghiêm trọng nào.
Polypeptide thiazolidic
Đại diện cho nhóm này là Bacitracin.
Bacitracin là một hỗn hợp peptide vòng lần đầu thu được từ chủng Tracy của Bacillus subtilis năm 1943.
Phổ tác dụng của nó là trên các vi khuẩn gram dương.
Cơ chế tác dụng của Bacitracin là ức chế tổng hợp thành tế bào, thông qua ức chế dephosphoryl hóa, ngăn cản sự chuyển các tiểu đơn vị peptidoglycan vào thành tế bào đang phát triển. Cơ chế tác dụng này là độc nhất so với các nhóm kháng sinh khác, do vậy không có sự đề kháng chéo giữa Bacitracin với các kháng sinh khác.
Bacitracin chỉ được sử dụng tại chỗ, không thể sử dụng đường toàn thân vì độc tính trên thận cao. Khả năng hấp thu của thuốc qua đường tiêu hóa cũng rất kém do kích thước phân tử lớn.
Bacitracin thường được kết hợp với Polymyxin hoặc Neomycin để điều trị nhiễm trùng trên bề mặt ở da hoặc niêm mạc. Không dùng trên vết thương hở do nguy cơ gặp phản ứng quá mẫn.
Kháng sinh nhóm Quinolone
Đây là nhóm kháng sinh có nguồn gốc từ tổng hợp hóa học hoàn toàn. Tất cả các Quinolone được sử dụng nhiều trên lâm sàng đều là dẫn chất của Nalidixic acid, và đa số có gắn nguyên tử fluor (F), gọi là các Fluoroquinolone.
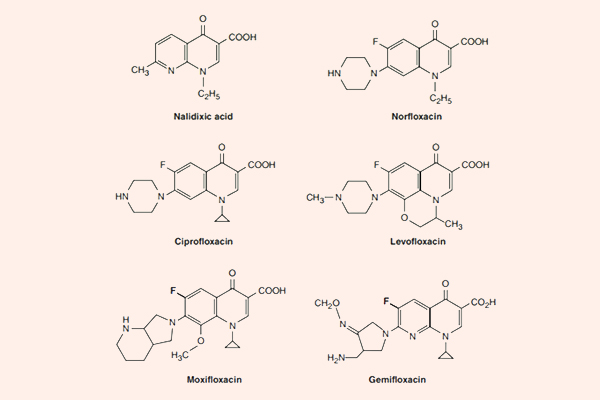
Cơ chế tác dụng
Các kháng sinh nhóm này ức chế tổng hợp ADN vi khuẩn bằng cách ức chế 2 enzyme topoisomerase II (ADN gyrase) and topoisomerase IV (thế hệ 1 chỉ ức chế topoisomerase II). Ức chế ADN gyrase làm ngăn cản quá trình tháo xoắn của ADN, điều này là cần thiết cho sự khởi đầu sao chép và phiên mã. Ức chế topoisomerase IV cản trở sự phân tách ADN nhiễm sắc thể sau sao chép vào các tế bào con tương ứng khi phân chia tế bào.
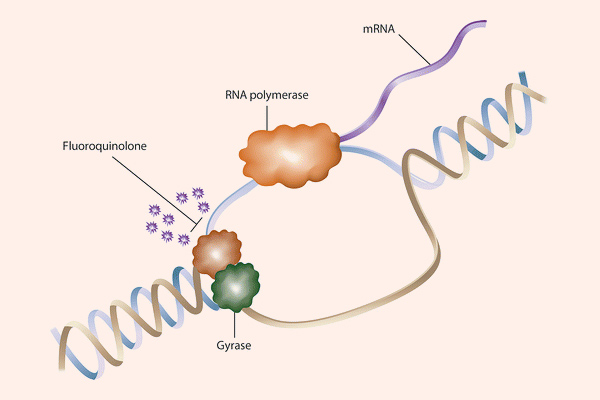
Phổ kháng khuẩn
Phổ kháng khuẩn được dùng để phân loại các thế hệ Quinolone. Hiện tại Quinolone được chia thành 4 thế hệ như được nói ở dưới.
Cơ chế kháng thuốc
Các cơ chế đề kháng với kháng sinh nhóm Quinolone đã được nghiên cứu bao gồm: Thay đổi đích tác dụng của kháng sinh (ADN gyrase và topoisomerase IV), thay đổi tính thấm của màng tế bào vi khuẩn với kháng sinh, hoặc tăng cường biểu hiện các bơm tống thuốc. Khả năng đề kháng chéo giữa các kháng sinh cùng nhóm là không cao.
Dược động học
Hấp thu: Các Quinolone nhìn chung có hấp thu qua đường tiêu hóa tốt và sinh khả dụng đường uống cao (80-95%). Hấp thu cũng như sinh khả dụng của các Quinolone bị giảm rất nhiều nếu dùng cùng các chế phẩm có chứa cation kim loại hóa trị II và III như nhôm, magnesi, calcium, sắt do chúng tạo phức chelate với thuốc. Nên dùng Quinolone 2 giờ trước hoặc 4 giờ sau khi sử dụng các sản phẩm chứa các cation kim loại này (điển hình là các Antacid).
Phân bố: Phân bố tốt vào các dịch và mô trong cơ thể.
Thải trừ: Thời gian bán thải nhìn chung thay đổi giữa các kháng sinh cụ thể (3-10 giờ). Levofloxacin, Gemifloxacin và Moxifloxacin có thể dùng 1 lần/ngày. Hầu hết các Quinolone được thải trừ ở thận thông qua cơ chế lọc ở cầu thận và/hoặc bài tiết ở ống thận và cần hiệu chỉnh liều ở bệnh nhân suy giảm chức năng thận. Nhưng Moxifloxacin là 1 trường hợp đặc biệt. Nó được thải trừ chủ yếu qua gan, nên không cần hiệu chỉnh liều ở bệnh nhân suy giảm chức năng thận, nhưng cần thận trọng ở bệnh nhân suy giảm chức năng gan.
Chỉ định
Các Fluoroquinolone được chỉ định cho nhiễm trùng đường tiết niệu (ngoại trừ Moxifloxacin), bao gồm cả do trực khuẩn mủ xanh gây ra, tiêu chảy do vi khuẩn lỵ, thương hàn, E.coli và Campylobacter, nhiễm trùng da và mô mềm, nhiễm trùng xương khớp, nhiễm trùng ổ bụng, nhiễm trùng hô hấp (các Quinolone thế hệ 3 và 4), bao gồm cả nguyên nhân do trực khuẩn mủ xanh. Một số chỉ định khác bao gồm bệnh than (Ciprofloxacin), bệnh lậu (Gemifloxacin), viêm cổ tử cung và viêm niệu đạo do Chlamydia (Levofloxacin và Ofloxacin), lao non-tuberculosis (Ciprofloxacin, Levofloxacin và Moxifloxacin), viêm màng não do não mô cầu…
Các kháng sinh nhóm Quinolone là kháng sinh dự trữ trong đa số các trường hợp, chỉ dùng khi mà không có thuốc khác tốt hơn.
Tác dụng không mong muốn
Rối loạn tiêu hóa: Buồn nôn, nôn và tiêu chảy.
Rối loạn thần kinh: Đau đầu, chóng mặt, loạn thần…, đặc biệt khi dùng liều cao.
Nhạy cảm ánh sáng: Do cơ chế hình thành các gốc tự do của thuốc.
Tim mạch: Kéo dài khoảng QT trên ECG. Tránh hoặc thận trọng khi sử dụng các thuốc này trên bệnh nhân có nguy cơ kéo dài khoảng QT, hạ kali máu hoặc dùng cùng thuốc khác cũng có khả năng gây kéo dài khoảng QT như thuốc chống loạn nhịp nhóm IA (Quinidine, Procainamide…), nhóm III (Sotalol, Ibutilide, Amiodarone…), các kháng sinh nhóm Macrolide (Erythromycin, Clarithromycin, Azithromycin).
Gatifloxacin đã bị rút khỏi thị trường Hoa Kỳ năm 2006 do gây tăng đường huyết ở bệnh nhân đái tháo đường và hạ đường huyết ở bệnh nhân dùng thuốc hạ đường huyết đường uống (đã có case tử vong).
Không khuyến cáo sử dụng Fluoroquinolones cho trẻ em dưới 18 tuổi do các nghiên cứu trên động vật cho thấy nó ảnh hưởng đến sự phát triển của sụn của động vật còn non. Tuy nhiên đây vẫn chỉ là nguy cơ mang tính lý thuyết và vẫn có những trường hợp bác sĩ sử dụng thuốc nhóm này cho trẻ em dưới 18 tuổi, đặc biệt là khi không có kháng sinh khác tốt hơn.
Các Fluoroquinolones cũng gây viêm gân, đứt gân (đứt gân ít gặp hơn), đặc biệt là gân achilles. Nguy cơ này có khả năng xảy ra cao hơn ở người cao tuổi, suy thận, hoặc có sử dụng corticoid đồng thời.
Không sử dụng thuốc cho phụ nữ có thai.
Quinolone thế hệ 1
Các kháng sinh thuộc phân nhóm này có hoạt lực trung bình, phổ hẹp, chỉ tác dụng trên vi khuẩn gram âm, trừ trực khuẩn mủ xanh. Đại diện tiêu biểu là Nalidixic acid. Hiện nay các nhóm thuốc này bị kháng rất nhiều, nên chúng chỉ được phê duyệt cho nhiễm trùng đường tiết niệu dưới không biến chứng.
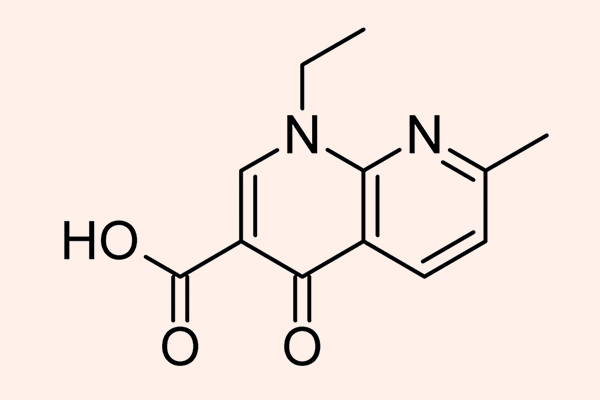
Quinolone thế hệ 2
Các thuốc thuộc nhóm 2 này đã mở rộng phổ trên cả vi khuẩn gram âm và gram dương, trong đó đã có thuốc có tác dụng tốt trên trực khuẩn mủ xanh (tốt nhất là Ciprofloxacin), phổ trên vi khuẩn gram dương bao gồm tụ cầu và liên cầu, trừ MRSA. Tác dụng trên phế cầu không đáng tin cậy, vì thế nên các thuốc thế hệ này không nên được sử dụng cho điều trị kháng sinh theo kinh nghiệm cho nhiễm trùng đường hô hấp. Các vi khuẩn không điển hình cũng nằm trong phổ tác dụng của các kháng sinh này. Các thuốc thế hệ này không có tác dụng trên vi khuẩn kỵ khí và các cầu khuẩn ruột gram dương.
Các thuốc trong phân nhóm này có thể được sử dụng cho nhiễm trùng đường tiết niệu có biến chứng, nhiễm trùng xương khớp, nhiễm trùng huyết, bệnh than…
Quinolone thế hệ 3
Các thuốc thế hệ 3 này còn gọi là Quinolone hô hấp, vì chúng có đã có tác dụng tốt trên phế cầu và có thể sử dụng để điều trị nhiễm trùng hô hấp. Hoạt tính của các thuốc nhóm này trên tụ cầu cũng tốt hơn thế hệ 2, nhưng hoạt tính trên vi khuẩn gram âm không tốt bằng thế hệ 2. Không có tác dụng trên vi khuẩn kị khí.
Các đại diện trong nhóm là Levofloxacin, Gatifloxacin, Gemifloxacin. Chúng thường được chỉ định (đặc biệt là Levofloxacin) trong nhiễm trùng hô hấp dưới. Ngoài ra chúng cũng có thể dùng trong nhiễm khuẩn đường tiết niệu, nhiễm khuẩn huyết…
Quinolone thế hệ 4
Đại diện duy nhất trong nhóm này là Moxifloxacin. Một số tài liệu có thể xếp Moxifloxacin là thế hệ 3, còn thế hệ 4 là Alatrofloxacin và Trovafloxacin. Tuy nhiên hiện tại 2 thuốc này đã bị dừng lưu hành từ năm 2006 do độc tính trên gan nghiêm trọng. Do vậy bài viết này sẽ xếp Moxifloxacin vào thế hệ 4.
Ưu điểm của Moxifloxacin là có phổ tác dụng mở rộng trên vi khuẩn kị khí, do đó nó có thể được dùng cho điều trị nhiễm khuẩn ổ bụng, bệnh viêm vùng chậu… Thuốc không được chỉ định cho nhiễm khuẩn đường tiết niệu vì thải trừ chủ yếu qua mật, thay vào đó có thể sử dụng nó cho nhiễm trùng đường mật. Hoạt tính trên trực khuẩn mủ xanh kém.
Kháng sinh nhóm Sulfamide và Trimethoprim
Các kháng sinh trong nhóm này đều có cơ chế ức chế tổng hợp acid folic của vi khuẩn. Do vậy chúng còn được gọi là nhóm kháng sinh kháng chuyển hóa.
p-aminobenzoic acid (PABA) là nguyên liệu quan trọng cho tổng hợp acid folic của vi khuẩn. Cấu trúc hóa học cơ bản của các Sulfamide đều bắt chước PABA. Các kháng sinh nhóm này có thể tan cả trong dung dịch có pH acid hoặc kiềm, nhưng tan trong pH kiềm nhiều hơn.
Cơ chế tác dụng
Các tế bào vi khuẩn nhìn chung không thể sử dụng folate ngoại sinh mà phải tổng hợp nó từ PABA. Acid folic cần thiết cho tổng hợp acid nucleic của vi khuẩn. Do các Sulfamide có cấu trúc học học tương tự PABA, nên chúng sẽ ức chế cạnh tranh enzyme dihydropteroate synthase, gây ức chế tổng hợp folate. Do cơ chế như vậy, phổ tác dụng các kháng sinh này rất rộng, bao gồm nhiều vi khuẩn gram dương và gram âm, ví dụ như tụ cầu vàng, E.coli, K.pneumoniae, trực khuẩn lỵ, thương hàn, Enterobacter sp., Nocardia sp., C.trachomatis, đồng thời cả một số động vật nguyên sinh. Thuốc hầu như không có tác dụng trên vi khuẩn kỵ khí, Rickettsiae và trực khuẩn mủ xanh.
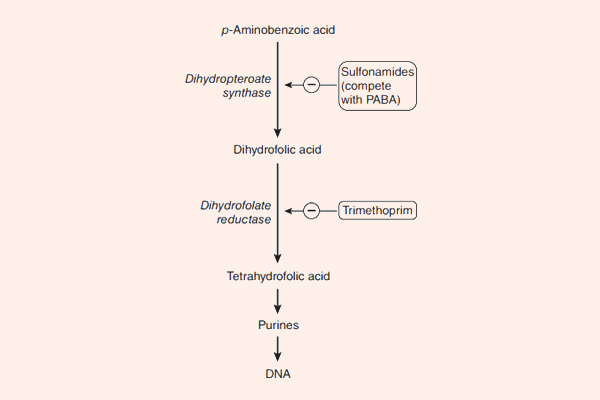
Các kháng sinh nhóm Sulfamide thường được phối hợp với các thuốc ức chế dihydrofolate reductase (bao gồm Trimethoprim hoặc Pyrimethamine) để tạo ra hiệp đồng tác dụng ức chế tổng hợp folate, tăng hiệu quả điều trị và giảm nguy cơ đề kháng thuốc.
Hiện nay, tỷ lệ đề kháng với các kháng sinh nhóm này đang tăng cao.
Cơ chế đề kháng
Các cơ chế đề kháng với Sulfamide đã được phát hiện bao gồm: Vi khuẩn thích nghi với sử dụng folate ngoại sinh, vi khuẩn thiếu enzyme tổng hợp folate từ PABA, sản xuất quá nhiều PABA, thay đổi enzyme tổng hợp folate làm giảm ái lực với thuốc, hoặc giảm tính thấm màng tế bào với thuốc. Nếu vi khuẩn kháng với 1 Sulfamide, nó sẽ kháng với tất cả kháng sinh trong nhóm.
Các cơ chế đề kháng với Trimethoprim gần tương tự, với các cơ chế giảm tính thấm màng tế bào với thuốc, sản xuất quá mức dihydrofolate reductase, hoặc thay đổi enzyme làm giảm ái lực liên kết với thuốc.
Dược động học
Hấp thu: Các Sulfamide dùng đường tại chỗ không hấp thu. Các Sulfamide dùng đường uống hấp thu tốt.
Phân bố: Nếu uống, các thuốc trong nhóm phân bố tốt vào các mô và dịch trong cơ thể, bao gồm cả hệ thần kinh trung ương và dịch não tủy. Thuốc qua được hàng rào nhau thai.
Chuyển hóa: Thông qua acetyl hóa hoặc glucuronide hóa ở gan.
Thải trừ: Chủ yếu ở thận theo cơ chế lọc ở cầu thận. Có thể cần hiệu chỉnh liều thuốc ở bệnh nhân suy giảm chức năng thận.
Trimethoprim/Sulfamethoxazole được phối hợp với nhau theo tỷ lệ liều 1:5 để tạo ra nồng độ đỉnh trong huyết thanh khoảng 1:20, đây là tỷ lệ cho tác dụng tối đa in vitro.
Trimethoprim cũng thấm tốt vào dịch tuyến tiền liệt và dịch âm đạo hơn các kháng sinh khác. Do vậy, nó có thể được dùng cho nhiễm khuẩn ở các cơ quan này.
Chỉ định
Sulfamide ít khi được sử dụng đơn độc do tình trạng kháng thuốc.
Sulfadiazine/Pyrimethamine chỉ định đầu tay cho bệnh do Toxoplasma cấp.
Sulfadoxine/Pyrimethamine chỉ định bậc hai cho điều trị sốt rét. Pyrimethamine có tác dụng phụ gây thiếu máu do ức chế tổng hợp folate, hạn chế tác dụng không mong muốn này bằng cách sử dụng Leucovorin.
Sulfacetamide nhãn khoa chỉ định cho điều trị viêm kết mạc nhiễm khuẩn hoặc hỗ trợ trong điều trị đau mắt hột.
Sulfadiazine bạc chỉ định cho sự phòng nhiễm trùng vết bỏng.
Trimethoprim/Sulfamethoxazole chỉ định cho điều trị viêm phổi do Pneumocystis jiroveci (trước đây là P.carinii), bệnh do Toxoplasma, hoặc Nocardia. Các chỉ định khác là nhiễm trùng đường tiết niệu, viêm tuyến tiền liệt, bệnh lỵ và thương hàn, nhiễm trùng hô hấp, nhiễm trùng da và mô mềm không biến chứng, nhiễm trùng xương khớp, nhiễm khuẩn huyết (không ưu tiên), nhiễm trùng Listeria (chỉ sử dụng thay thế khi bệnh nhân không dung nạp Ampicillin). Hiện nay tỷ lệ các chủng vi khuẩn kháng thuốc ngày càng tăng cao, làm việc chỉ định thuốc để điều trị kháng sinh theo kinh nghiệm bị giới hạn nhiều hơn.
Tác dụng không mong muốn
Có phản ứng dị ứng chéo giữa các bệnh nhân dùng Sulfamide với Diazoxide và các Sulfonylurea hạ đường huyết do cấu trúc hóa học tương đồng. Thật may là điều này không phổ biến.
Các phản ứng dị ứng khác có thể gặp với thuốc là phát ban, viêm da tróc vảy, mày đay, nặng có hội chứng Stevens-Johnson (tần suất dưới 1%).
Rối loạn tiêu hóa: Buồn nôn, nôn và tiêu chảy.
Thuốc có thể gây sỏi đường tiết niệu. Nguyên nhân hình thành sỏi đường tiết niệu là do các Sulfamide hoặc sản phẩm chuyển hóa của nó kết tủa ở pH trung tính hoặc hơi acid của nước tiểu. Uống nhiều nước có thể giảm thiểu tác dụng không mong muốn này.
Các Sulfamide cũng có tác dụng không mong muốn trên hệ tạo máu: Thiếu máu tan huyết, thiếu máu bất sản, giảm bạch cầu hạt, giảm tiểu cầu. Phản ứng tan máu dễ xảy ra ở bệnh nhân thiếu G6PD (Glucose-6-phosphate Dehydrogenase).
Nếu sử dụng các Sulfamide vào các tháng cuối thai kỳ có thể gây ra vàng da nhân não ở trẻ sơ sinh.
Trimethoprim có các tác dụng không mong muốn là gây ra thiếu máu hồng cầu khổng lồ (do ức chế tổng hợp acid folic), giảm bạch cầu hạt.
Kháng sinh nhóm 5-nitroimidazole
Đây là nhóm kháng sinh tổng hợp hoàn toàn, với các đại diện là Metronidazole, Tinidazole, Ornidazole và Secnidazole. Ban đầu các kháng sinh nhóm này ra đời để điều trị nhiễm trùng các động vật nguyên sinh, trong đó có amip. Sau này, người ta mới phát hiện ra rằng nó tác dụng tốt trên nhiều vi khuẩn kị khí.
Các kháng sinh nhóm này có tác dụng tốt trên các vi khuẩn kị khí chủng Bacteroides và Clostridium. Ngay sau khi vào trong tế bào vi khuẩn kị khí, nó bị khử theo con đường không enzyme bằng phản ứng với ferredoxin khử. Nhóm nitro (-NO2) của các kháng sinh nhóm này chuyển thành nhóm nitroso (-NO) rất độc với các thành phần tế bào, đặc biệt là ADN. ADN bị tổn thương làm chết tế bào vi khuẩn. Trong các vi khuẩn hiếu khí, sự khử không xảy ra, mà nếu có xảy ra thì nhóm nitroso cũng nhanh chóng bị oxy hóa về nhóm nitro. Do vậy, các vi khuẩn hiếu khí đề kháng tự nhiên với các kháng sinh nhóm này.
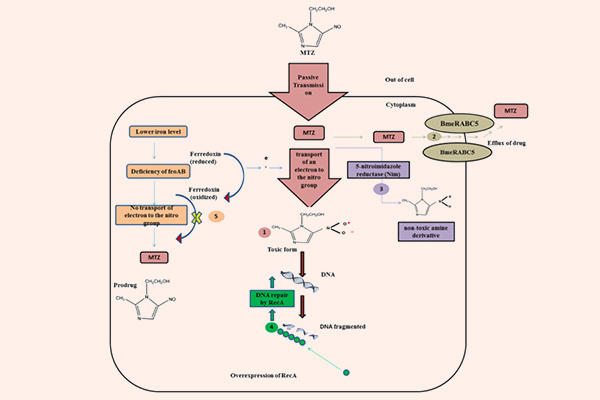
Các kháng sinh nhóm này hấp thu tốt qua đường tiêu hóa và có sinh khả dụng đường uống cao. Chúng phân bố rộng vào các mô, vào cả não và dịch não tủy, với nồng độ tương đương nồng độ trong huyết thanh. Thuốc được chuyển hóa ở gan. Các thuốc trong nhóm chỉ khác nhau về dược động học, chủ yếu ở thời gian bán thải (Metronidazole có thời gian bán thải ngắn nhất), còn dược lực học tương tự nhau.
Thuốc được chỉ định cho nhiễm trùng kỵ khí hoặc hỗn hợp trong ổ bụng (kết hợp với các kháng sinh khác chống vi khuẩn hiếu khí), viêm âm đạo (do vi khuẩn hoặc Trichomonas), nhiễm trùng amip (trong lòng ruột hoặc áp xe gan do amip), nhiễm trùng Giardia và viêm đại tràng giả mạc do C.difficile.
Các tác dụng không mong muốn trên hệ tiêu hóa là buồn nôn, tiêu chảy, lưỡi có vị kim loại. Sử dụng thuốc kéo dài có thể gây ra bệnh lý thần kinh ngoại biên. Các thuốc nhóm này cũng có phản ứng giống Disulfiram khi sử dụng đồ uống có cồn, do đó khi đang dùng thuốc thì tuyệt đối không dùng đồ uống hoặc thức ăn có chứa cồn.
Kháng sinh nhóm Pleuromutilin
So với nhiều nhóm kháng sinh đã được kể ở trên, thì nhóm kháng sinh này có lẽ sẽ còn hơi xa lạ với nhiều người. Hiện nay, các kháng sinh nhóm này chưa được đưa vào giảng dạy trong các chương trình Dược lý đại học, hoặc có thể đã được đưa vào nhưng nội dung giảng dạy còn nhiều hạn chế. Nguyên do là bởi hầu như tất cả các kháng sinh nhóm này đều chỉ được sử dụng cho vật nuôi là chính, chứ không được sử dụng trên người. Thêm vào đó, kháng sinh duy nhất được dùng trên người theo đường toàn thân của nhóm này là Lefamulin, cũng chỉ vừa mới được FDA cấp phép không lâu (tháng 8/2019).
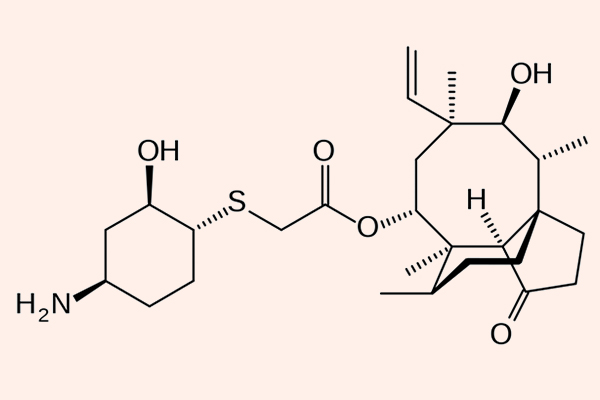
Lefamulin ức chế tổng hợp protein vi khuẩn bằng cách gắn và ức chế tiểu đơn vị ribosome 50S của vi khuẩn. Cơ chế của nó hoàn toàn khác với các kháng sinh ức chế tiểu đơn vị ribosome 50S khác như các Macrolide, Lincosamide, Streptogramin, Phenicol, Oxazolidinone. Không có kháng chéo giữa Lefamulin với các nhóm kháng sinh khác.
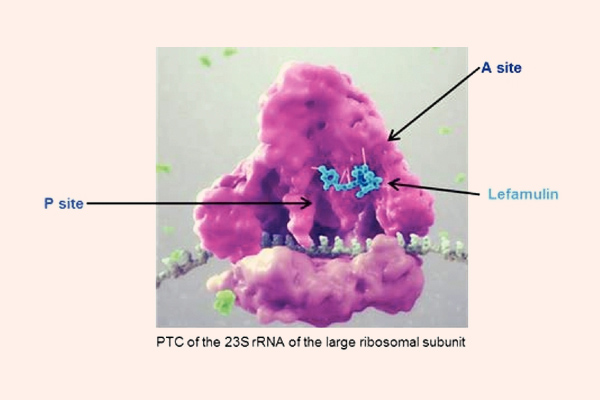
Lefamulin được FDA phê duyệt cho viêm phổi mắc phải tại cộng đồng do phế cầu, tụ cầu vàng nhạy cảm Methicillin, H.influenzae, L.pneumophila, M.pneumoniae và C.pneumoniae. Tuy nhiên, thuốc đã thể hiện hoạt tính in vitro trên MRSA, E.faecium kháng Vancomycin và lậu đa kháng thuốc. Không cần hiệu chỉnh liều trong suy giảm chức năng thận, nhưng cần hiệu chỉnh liều trong bệnh gan tiến triển.
Các tác dụng không mong muốn phổ biến là buồn nôn, nôn, tiêu chảy, viêm gan và kéo dài khoảng QT trên ECG.
Kháng sinh chống lao (M.tuberculosis)
Lao là bệnh bắt buộc phải phối hợp thuốc điều trị và phải điều trị trong một thời gian dài. Vi khuẩn lao rất dễ kháng thuốc.
Kháng sinh đầu tay
Kháng sinh đầu tay điều trị lao gồm 5 thuốc: S (Streptomycin), R (Rifampin hoặc các Ryfamycin khác), H (Isoniazid), Z (Pyrazinamide) và E (Ethambutol). Trong đó nhiều nước đã rút S khỏi danh sách các thuốc đầu tay điều trị lao. Trong các thuốc trên thì H và R là 2 thuốc có hoạt tính tốt nhất.
Isoniazid
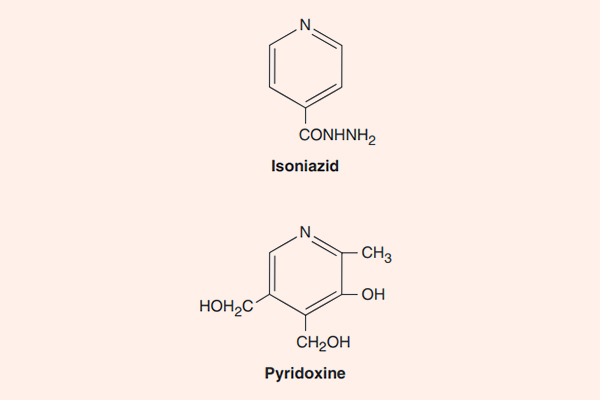
Isoniazid là thuốc có hoạt tính cao nhất trên trực khuẩn lao M.tuberculosis. Nó là một phân tử có cấu trúc hóa học tương tự vitamin B6. Isoniazid xâm nhập tốt vào đại thực bào nên có tác dụng trên cả các vi khuẩn lao ngoại bào hoặc nội bào.
Cơ chế tác dụng của Isoniazid là ức chế tổng hợp acid mycolic, đây là thành phần thiết yếu trong vách tế bào vi khuẩn lao. Isoniazid là một tiền thuốc (prodrug) và được hoạt hóa bởi enzyme KatG của vi khuẩn lao. Isoniazid sau khi được hoạt hóa sẽ hình thành liên kết cộng hóa trị với AcpM và KasA, điều này làm ức chế tổng hợp acid mycolic.
Cơ chế đề kháng với Isoniazid của trực khuẩn lao có liên quan đến biểu hiện quá mức inhA (kháng chéo với Ethionamide) hoặc ahpC (liên quan đến đột biến KasA), hay đột biến thay đổi hoặc loại bỏ gen quy định katG.
Về dược động học, Isoniazid được hấp thu nhanh qua đường tiêu hóa và đạt tối đa khi đói. Bữa ăn chứa nhiều chất béo có thể làm giảm nồng độ đỉnh trong huyết tương của thuốc tới 50% nếu dùng cùng. Phân bố tốt vào các mô và dịch trong cơ thể. Thuốc có thể vào được dịch não tủy. Ở gan, thuốc được chuyển hóa nhờ phản ứng acetyl hóa bởi N-acetyltransferase. Sự chuyển hóa này là khác nhau ở mỗi người (tính đa hình di truyền). Có người mang kiểu hình acetyl hóa nhanh sẽ cần dùng thuốc với liều cao hơn, nhưng những người mang kiểu hình acetyl hóa chậm sẽ lại cần dùng thuốc với liều thấp hơn. Thải trừ thuốc và các chất chuyển hóa của nó chủ yếu là qua nước tiểu.
Isoniazid có tương tác thuốc với một số thuốc như Phenytoin, Carbamazepine và các Benzodiazepine. Nồng độ các thuốc này trong huyết tương tăng lên khi dùng cùng Isoniazid do Isoniazid ức chế một số enzyme chuyển hóa thuốc trong gan.
Các tác dụng không mong muốn bao gồm: Sốt, phát ban, lupus ban đỏ hệ thống do thuốc, độc tính trên gan (tăng men gan, viêm gan) và bệnh lý thần kinh ngoại biên. Ngoài ra còn một số tác dụng không mong muốn khác. Độc tính trên gan cần chú ý theo dõi bệnh nhân chặt chẽ và liên tục trong quá trình điều trị và cần thận trọng ở những người có yếu tố nguy cơ cao. Bệnh lý thần kinh ngoại biên có liên quan đến thiếu vitamin B6 do sự tương đồng trong cấu trúc hóa học giữa thuốc và vitamin B6. Bổ sung vitamin B6 liều thấp hàng ngày là cần thiết.
Rifampin
Rifampin là một dẫn chất bán tổng hợp từ Rifamycin, một kháng sinh có nguồn gốc từ Amycolatopsis rifamycinica, trước đây có tên khoa học là Streptomyces mediterranei. Nó có phổ tác dụng trên vi khuẩn gram dương và một số vi khuẩn gram âm, như Neisseria, Haemophilus và Chlamydia. Có sự đề kháng chéo giữa các dẫn chất của Rifamycin với nhau.
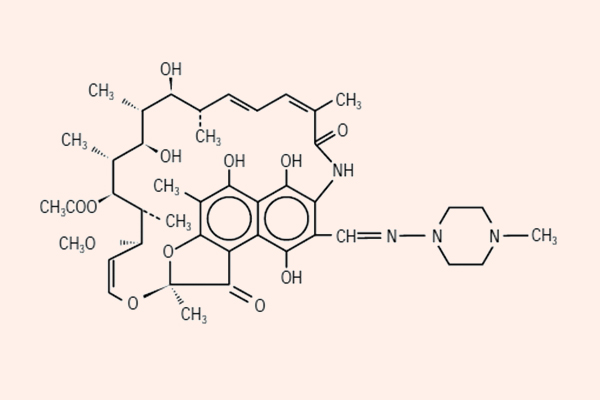
Cơ chế tác dụng của Rifampin và các dẫn chất của Rifamycin nói chung là ức chế tổng hợp ARN do ức chế ARN polymerase phụ thuộc ADN của vi khuẩn. Cơ chế đề kháng thuốc chính là thay đổi đích tác dụng ARN polymerase của kháng sinh, thông qua đột biến điểm rpoB. Rifampin là kháng sinh diệt khuẩn đối với vi khuẩn lao. Thuốc này cũng có thể xâm nhập vào đại thực bào như Isoniazid.
Dược động học: Thuốc được hấp thu tốt qua đường uống. Phân bố rộng vào các mô và dịch trong cơ thể, liên kết với protein huyết tương cao, vào được dịch não tủy, vào nhiều hơn khi màng não bị viêm. Nó được chuyển hóa tại gan và có chu kỳ gan – ruột. Thải trừ chủ yếu qua mật.
Do là chất cảm ứng mạnh CYP450 của gan (CYP1A2, CYP2C9, CYP2C19, CYP2D6 và CYP3A4), tương tác thuốc rất dễ xảy ra. Nó có thể làm giảm nồng độ nhiều thuốc trong huyết tương nếu dùng cùng.
Ngoài chỉ định cho lao, thuốc còn nhiều chỉ định khác, nhưng ta không quan tâm tới ở đây.
Rifampin làm cho nước tiểu, mồ hôi và nước mắt có màu cam. Cần chú ý điều này.
Thuốc cũng có độc tính trên gan, gây vàng da ứ mật hoặc viêm gan. Theo dõi chức năng gan liên tục trong suốt quá trình điều trị.
Ethambutol
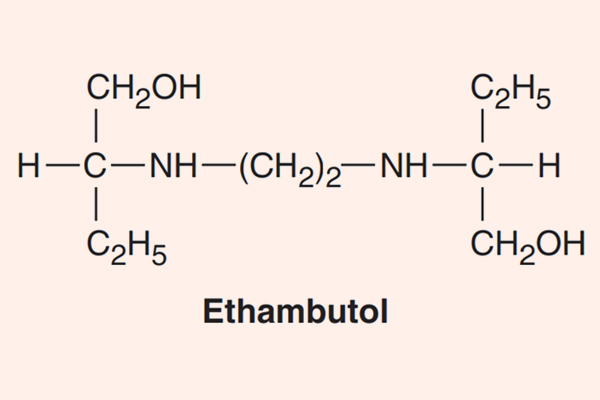
Cơ chế tác dụng của Ethambutol là ức chế arabinosyl transferase của vi khuẩn lao, từ đó ức chế tổng hợp thành tế bào vi khuẩn. Arabinosyl transferase tham gia phản ứng polymerase hóa arabinoglycan, đây là một thành phần thiết yếu của thành tế bào vi khuẩn lao. Cơ chế đề kháng với thuốc thường là thay đổi đích tác dụng của thuốc.
Dược động học: Ethambutol được hấp thu tốt qua đường tiêu hóa. Thuốc chỉ đi qua hàng rào máu não chỉ khi màng não bị viêm. Thải trừ thuốc qua cả phân (20%) và nước tiểu (50%). Cần hiệu chỉnh liều ở bệnh nhân suy giảm chức năng thận.
Tác dụng không mong muốn nổi bật của thuốc là viêm dây thần kinh thị giác (sau nhãn cầu), gây ra hậu quả là mất thị lực và mù màu xanh-đỏ. Chống chỉ định tương đối ở trẻ em.
Pyrazinamide
Pyrazinamide bị bất hoạt ở pH trung tính, nhưng ở pH 5.5, nó có thể ức chế trực khuẩn lao. Thuốc có thể được hấp thu vào đại thực bào và tiêu diệt vi khuẩn lao cư trú trong các lysosome vốn có môi trường acid. Do đó thuốc tiêu diệt các vi khuẩn lao còn sót lại sau đợt điều trị tấn công trong phác đồ điều trị lao.
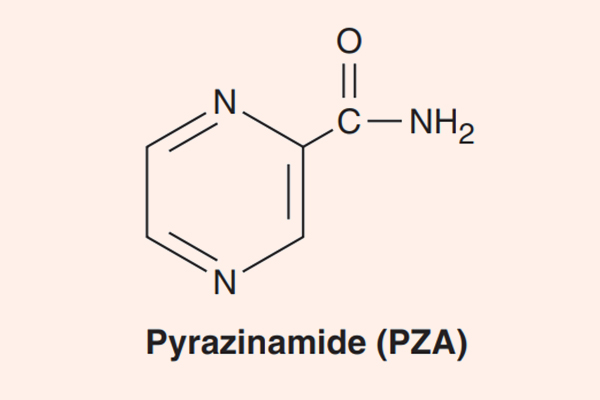
Pyrazinamide là tiền thuốc, nó phải được chuyển thành dạng hoạt hóa của nó là Pyrazinoic acid nhờ một enzyme của trực khuẩn lao có tên là pyrazinamidase. Pyrazinoic acid ức chế chuyển hóa màng tế bào vi khuẩn lao. Vi khuẩn lao có thể kháng với thuốc bằng cách giảm tính thấm màng vi khuẩn với tế bào, hoặc đột biến enzyme pyrazinamidase, làm giảm chuyển Pyrazinamide thành dạng có hoạt tính Pyrazinoic acid.
Dược động học: Thuốc được hấp thu nhanh qua đường tiêu hóa. Phân bố tốt vào các mô trong cơ thể, có thể đi vào dịch não tủy nếu màng não bị viêm. Chuyển hóa thuốc ở gan. Thời gian bán thải khoảng 8-11 giờ. Thải trừ chủ yếu qua thận và cần hiệu chỉnh liều ở bệnh nhân suy giảm chức năng thận.
Thuốc có độc tính trên gan như ít hơn Isoniazid và Rifampin. Các tác dụng không mong muốn khác là rối loạn tiêu hóa với buồn nồn và nôn, nhạy cảm ánh sáng, tăng acid uric máu (thận trọng với bệnh nhân mắc bệnh gout)…
Streptomycin
Xem phần kháng sinh nhóm Aminoside.
Kháng sinh bậc hai
Ethionamide
Ethionamide có cấu trúc tương tự Isoniazid và cơ chế tác dụng cũng tương tự. Giữa 2 thuốc này có thể có sự đề kháng chéo của vi khuẩn lao.
Dược động học: Thuốc hấp thu tốt qua đường uống. Phân bố của thuốc vào dịch não tủy tốt. Chuyển hóa thuốc tại gan.
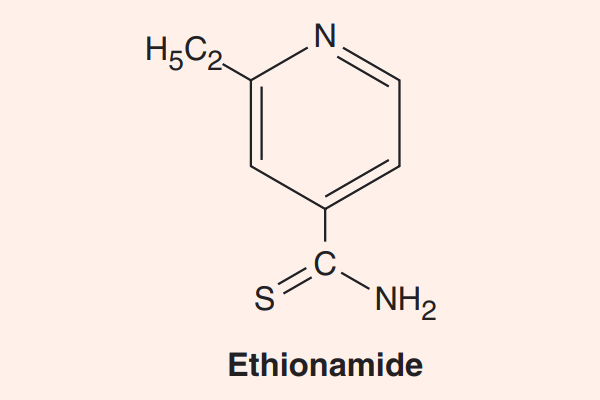
Thuốc cũng có độc tính trên gan và gây ra bệnh lý thần kinh ngoại biên tương tự như Isoniazid, cần bổ sung vitamin B6.
Capreomycin
Capreomycin là một kháng sinh peptide có nguồn gốc từ các chủng Streptomyces capreolus. Thuốc hoạt động theo cơ chế ức chế tổng hợp protein và phải dùng đường tiêm bắp tương tự như Streptomycin.
Capreomycin có độc tính trên thận và thính giác giống Streptomycin. Bệnh nhân có thể bị ù tai, điếc, rối loạn tiền đình.
Chế độ dùng thuốc ngắt quãng có thể làm giảm đến tối thiểu độc tính.
Cycloserine
Cycloserine là một thuốc có cấu trúc hóa học tương tự D-Alanine, có cơ chế tác dụng theo kiểu ức chế tổng hợp thành tế bào.
Dược động học: Phân bố rộng vào mô, kể cả hệ thần kinh trung ương. Thải trừ chủ yếu qua thận, cần hiệu chỉnh liều thuốc ở bệnh nhân suy giảm chức năng thận.
Thuốc có tác dụng không mong muốn là gây ra bệnh lý thần kinh ngoại biên, cùng với rối loạn tâm thần như trầm cảm và loạn thần. Để giảm thiểu bệnh lý thần kinh ngoại biên gây ra do thuốc, nên bổ sung vitamin B6.
Aminosalicylic acid (PAS)
PAS là một thuốc kháng folate có hoạt phổ rộng trên hầu hết các chủng vi khuẩn lao. Nó có cấu trúc hóa học tương tự PABA và được cho là có cơ chế tác dụng tương tự các Sulfamide đã được trình bày ở trên.
Dược động học: Thuốc được phân bố rộng vào các mô và dịch trong cơ thể, trừ dịch não tủy. Chuyển hóa thuốc ở gan bằng phản ứng acetyl hóa và một số phản ứng khác. Thải trừ thuốc chủ yếu qua nước tiểu. Cần hiệu chỉnh liều thuốc ở bệnh nhân suy giảm chức năng thận.
Các tác dụng không mong muốn của thuốc là gây sỏi đường tiết niệu (do thuốc kết tủa ở pH hơi acid của nước tiểu), gây loét và xuất huyết dạ dày (có thể do ức chế COX-1 [cyclooxygenase-1] đường tiêu hóa do cấu trúc hóa học tương tự Aspirin). Các tác dụng không mong muốn khác của thuốc là dị ứng và quá mẫn, với biểu hiện sốt, phát ban, đau khớp, viêm gan…
Kanamycin và Amikacin
Xem phần kháng sinh nhóm Aminoside.
Fluoroquinolone
Xem phần kháng sinh nhóm Quinolone. 4 kháng sinh có thể sử dụng là Ciprofloxacin, Levofloxacin, Gatifloxacin và Moxifloxacin.
Linezolid
Xem phần kháng sinh nhóm Oxazolidinone.
Rifabutin, Rifapentine
Rifabutin và Rifapentine là dẫn chất của Rifamycin và có cơ chế tác dụng tương tự như Rifampin.
Bedaquiline
Bedaquiline là một diarylquinoline (2 hệ vòng thơm và 1 nhân quinoline), đây là thuốc đầu tiên có cơ chế hoạt động mới chống lại M.tuberculosis kể từ năm 1971.
Cơ chế tác dụng của Bedaquiline là ức chế adenosine 5′-triphosphate (ATP) synthase của vi khuẩn lao, enzyme này chịu trách nhiệm tổng hợp ATP – dạng năng lượng chính cho mọi tế bào sống. Thiếu ATP làm vi khuẩn bị tiêu diệt. Có quan sát thấy hoạt tính diệt khuẩn của thuốc trên mô hình chuột thí nghiệm. Có sự đề kháng chéo giữa Bedaquiline và Clofazimine (thuốc điều trị bệnh phong), thông qua cơ chế bơm tống đa thuốc MmpL5.
Dược động học: Thuốc hấp thu tốt qua đường tiêu hóa, đặc biệt khi dùng cùng bữa ăn giàu chất béo. Tỷ lệ liên kết với protein huyết tương rất cao (> 99%). Thuốc được chuyển hóa ở gan, chủ yếu thông qua hệ enzyme CYP450, với enzyme chủ lực là CYP3A4, điều này gây ra những nguy cơ về tương tác thuốc trên lâm sàng. Thải trừ thuốc chủ yếu qua phân. Thời gian bán thải rất dài, trung bình lên tới 5.5 tháng (thời gian bán thải tính cho cả Bedaquiline và chất chuyển hóa M2 của nó).
Các tác dụng không mong muốn của Bedaquiline phổ biến nhất là buồn nôn, đau khớp và đau đầu. Thuốc cũng có độc tính trên gan, đồng thời có độc tính trên tim, gây kéo dài khoảng QT trên ECG. Thận trọng ở những bệnh nhân có bất thường ở dẫn truyền trong tim, đang dùng các thuốc khác cũng có thể gây kéo dài khoảng QT.
Một số kháng sinh khác
Nitrofurantoin
Nitrofurantoin là một kháng sinh dùng trong điều trị nhiễm khuẩn đường tiết niệu phổ biến.
Phổ tác dụng rộng trên nhiều vi khuẩn gram dương và gram âm, trừ trực khuẩn mủ xanh và Proteus sp. Cơ chế tác dụng chính xác chưa rõ ràng. Nhưng thuốc có thể phản ứng với các enzyme khử của vi khuẩn, tạo ra các chất hóa học trung gian phá hủy ribosome, gây rối loạn tổng hợp protein, phá hủy ADN và ARN.
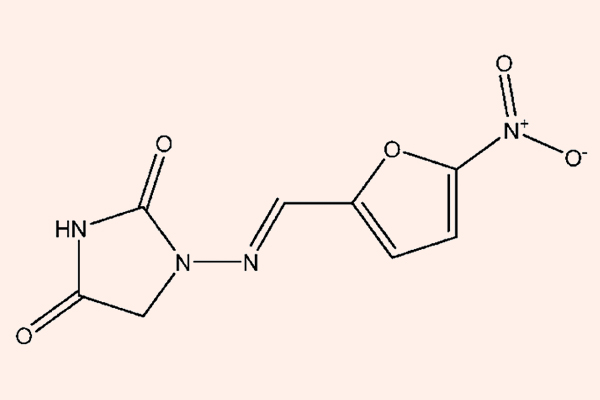
Nhờ cơ chế tác dụng độc nhất, không có sự đề kháng chéo giữa Nitrofurantoin với các nhóm kháng sinh khác.
Thuốc được chỉ định cho nhiễm trùng đường tiết niệu dưới không biến chứng.
Dược động học: Thuốc được hấp thu tốt bằng đường uống. Chuyển hóa và thải trừ rất nhanh qua thận (theo cơ chế lọc ở cầu thận và bài tiết ở ống thận) nên không có tác dụng kháng khuẩn toàn thân mà chỉ có tác dụng kháng khuẩn khi ở trong nước tiểu (pH nước tiểu dưới 5.5 làm tăng hoạt tính kháng khuẩn của thuốc). Chống chỉ định ở bệnh nhân có suy giảm chức năng thận đáng kể.
Các tác dụng không mong muốn chính của thuốc là rối loạn tiêu hóa, với chán ăn, buồn nôn và nôn. Các tác dụng không mong muốn khác ít gặp hơn là độc tính trên phổi, thần kinh, thiếu máu tan huyết ở bệnh nhân thiếu G6PD.
Fosfomycin
Fosfomycin là một kháng sinh tổng hợp hóa học toàn phần có cấu trúc đơn giản.
Thuốc ức chế tổng hợp thành tế bào, nhưng ở giai đoạn sớm hơn rất nhiều so với các β-lactam hoặc Glycopeptide. Fosfomycin bắt chước cấu trúc hóa học của phosphoenolpyruvate, ức chế không hồi phục enolpyruvate transferase (liên kết cộng hóa trị với acid amin cystein ở trung tâm hoạt động của enzyme), điều này làm cản trở sự bổ sung phosphoenolpyruvate vào UDP-N-acetylglucosamine. UDP-N-acetylglucosamine lại là chất cần thiết để tổng hợp UDP-N-acetylmuramic acid, từ chất này vi khuẩn mới tạo ra được N-acetylmuramic acid, thành phần quan trọng trong peptidoglycan ở thành tế bào vi khuẩn.
Để có tác dụng ức chế enzyme enolpyruvate transferase vốn ở trong tế bào chất, thuốc cần phải đi vào được trong tế bào. Ở vi khuẩn, hệ vận chuyển glycerophosphate hoặc glucose 6-phosphate chịu trách nhiệm cho sự vận chuyển thuốc. Do đó, vi khuẩn có thể đề kháng với Fosfomycin thông qua thay đổi hệ vận chuyển này, làm thuốc không vào được trong tế bào, hoặc vào nhưng đạt không đủ nồng độ cần thiết để ức chế tổng hợp thành tế bào vi khuẩn.
Hoạt phổ rộng với nhiều vi khuẩn gram dương và gram âm.
Dược động học: Thuốc có thể dùng đường uống hoặc tiêm. Sinh khả dụng đường uống khoảng 40%. Thải trừ thuốc nguyên vẹn qua nước tiểu, do đó thuốc rất thích hợp cho điều trị nhiễm trùng đường tiết niệu.
Fosfomycin được chỉ định cho điều trị nhiễm trùng đường tiết niệu (UTI) dưới không biến chứng ở phụ nữ. Hiệu lực trên nam giới bị thiếu dữ liệu, nhưng vẫn có thể sử dụng.
Methenamine
Methenamine có 2 dạng dược dụng hiện đang được sử dụng trên thị trường, đó là dạng muối mandelate (muối với acid mandelic) hoặc muối hippurate (muối với acid hippuric).
Thuốc được thải trừ nguyên vẹn qua nước tiểu. Ở pH < 5.5 trong nước tiểu, thuốc giải phóng ra formaldehyde (HCHO), đây là một chất sát khuẩn rất mạnh. pH nước tiểu cao hơn làm thuốc không có tác dụng.
Thuốc được chỉ định cho dự phòng nhiễm trùng đường tiết niệu có triệu chứng.
Có thể sử dụng thêm vitamin C để làm nước tiểu trở nên acid hơn, làm tăng hiệu lực của thuốc.
Không phối hợp thuốc này với các Sulfamide kháng khuẩn vì chúng có thể có phản ứng tạo tủa với formaldehyde được giải phóng từ Methenamine, làm giảm hiệu lực của thuốc.
Acid fusidic
Đây là một kháng sinh được sử dụng tại chỗ, sử dụng đường uống cũng có nhưng không phổ biến. Cấu trúc hóa học của nó khá đặc biệt khi có khung steroid trong đó.
Acid fusidic ức chế tổng hợp protein vi khuẩn, do đó có hoạt tính kìm khuẩn. Phổ tác dụng của nó chủ yếu giới hạn trên các chủng vi khuẩn gram dương như tụ cầu và liên cầu, bao gồm cả MRSA. Acid fusidic khi sử dụng đơn độc gây kháng thuốc nhanh.
Acid fusidic chủ yếu được sử dụng cho các nhiễm khuẩn ngoài da nhẹ và có thể dùng trong nhiễm trùng mắt.
Rifaximin
Đây mặc dù là dẫn chất của Rifampin và có cơ chế tác dụng cũng như phổ tác dụng tương tự Rifampin, nhưng chỉ định của nó lại khác hẳn.
Rifaximin hầu như không được hấp thu theo đường uống, vậy nên nó được dùng để điều trị tiêu chảy du lịch. Các chỉ định khác của thuốc bao gồm quản lý bệnh não gan, hội chứng ruột kích thích với tiêu chảy, và đôi khi là viêm đại tràng giả mạc do C.difficile tái phát hoặc dai dẳng.
Rifaximin hầu như không có tương tác thuốc qua hệ CYP450 ở gan do hấp thu qua đường tiêu hóa hạn chế.
Mupirocin
Mupirocin (pseudomonic acid) là một kháng sinh tự nhiên có nguồn gốc từ Pseudomonas fluorescens. Đây là kháng sinh được sử dụng tại chỗ.
Phổ tác dụng của Mupirocin chủ yếu bao gồm các vi khuẩn gram dương thường gây nhiễm trùng da, đó là tụ cầu vàng. Thuốc có tác dụng với cả các chủng đã đề kháng Methicillin (MRSA).
Cơ chế tác dụng của Mupirocin là ức chế một enzyme của tụ cầu có tên là isoleucyl-tARN synthetase. Đây là enzyme có tác dụng tổng hợp phức hợp isoleucyl-tARN. Khi không tổng hợp được nó, acid amin isoleucyl không thể gắn vào chuỗi peptide của vi khuẩn. Như vậy quá tình tổng hợp protein của vi khuẩn bị ức chế. Đây là kháng sinh kìm khuẩn ở nồng độ thấp, nhưng diệt khuẩn ở nồng độ cao.
Cơ chế đề kháng kháng sinh chính là thay đổi đích tác dụng của kháng sinh, thường do đột biến điểm trên gen trong nhiễm sắc thể, xảy ra khi sử dụng kéo dài. Nhưng may mắn là, nồng độ thuốc khi sử dụng tại chỗ rất cao, cao hơn MIC nhiều lần, nên cơ chế này thường không gây ra có thất bại điều trị. Tuy nhiên, nếu đột biến một gen khác quy định tổng hợp enzyme đích trên plasmid, vi khuẩn có thể kháng với thuốc kể cả ở nồng độ cao.
Mupirocin được chỉ định cho các nhiễm trùng da nhẹ, như chốc lở. Các nhiễm trùng lớn, vết thương phẫu thuật… không khuyến khích dùng thuốc.
Tài liệu tham khảo
Bertram G. Katzung, Mc Graw Hill Education, Basic and Clinical Pharmacology, 14th edition.
Đăng ký nhận tin
Nhập Email của bạn để nhận được thông tin hữu ích từ HealCentral.
Cam kết không Spam, chỉ gửi những thông tin sức khỏe có giá trị.
Cam kết không Spam, chỉ gửi những thông tin sức khỏe có giá trị.
Hỗ trợ
Fanpage

![]() © 2020 Bản quyền
HealCentral.org
© 2020 Bản quyền
HealCentral.org
Nội dung trên website đã được đăng ký bản quyền thiên niên kỷ DMCA, copy vui lòng để liên kết về bài viết.
Thông tin trên Web chỉ mang tính chất tham khảo. Dùng thuốc cần theo hướng dẫn của chuyên gia y tế. Tuyệt đối không tự sử dụng thuốc - Chúng tôi không chịu trách nhiệm về nội dung trên Web do thành viên đăng tải
0375.288.862