Amyloidosis là một tình trạng rối loạn rất hiếm gặp nhưng rất nguy hiểm, trong đó protein amyloid không được hòa tan lắng đọng trong các cơ quan cơ thể, gây ra sự tích tụ protein bất thường trong các mô và cuối cùng dẫn đến rối loạn chức năng nội tạng và tử vong. Để có thái độ xử trí đúng đắn, việc xác định chính xác protein là rất quan trọng vì tiên lượng và điều trị Amyloidosis có thể khác nhau tùy thuộc vào protein căn nguyên. Tuy nhiên Amyloidosis là những bệnh rất phức tạp để nghiên cứu và chẩn đoán vì chúng thường liên quan đến các hệ cơ quan khác nhau (trong trường hợp bệnh hệ thống) và các triệu chứng không rõ ràng có thể dễ dàng chẩn đoán sai. Bài viết sau đây Heal Central xin trình bày mô tả các cách phân loại và đặc điểm lâm sàng của amyloidosis và cung cấp cho bạn đọc một cái nhìn tổng quan về các công cụ chẩn đoán và phương pháp điều trị hiện tại.
Amyloidosis là gì?
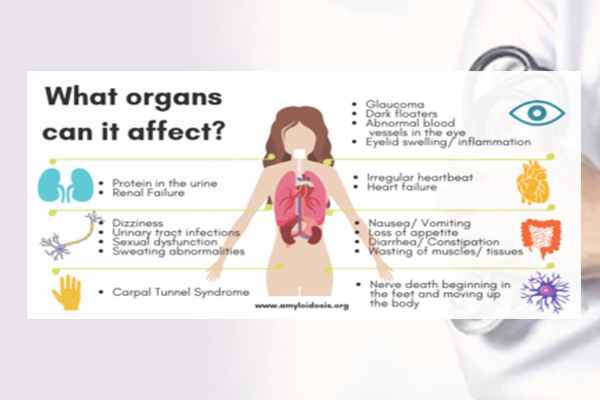
Amyloidosis không phải là một bệnh mà là tập hợp các bệnh do sự rối loạn về cấu trúc và chuyển hóa protein, trong đó các sợi cơ không hòa tan được lắng đọng trong các cơ quan cơ thể, gây ra rối loạn chức năng nội tạng và cuối cùng là tử vong. Người ta đã phân lập và xác định được khoảng 60 protein amyloidogen không đồng nhất, trong đó, 27 loại được ghi nhận gây bệnh ở người. Những rối loạn gây lên bất thường cấu trúc của các protein, sau đó hình thành các sợi cơ cứng không phân nhánh ức chế sự phân giải protein và gây ra sự gián đoạn cơ học và stress oxy hóa cục bộ ở các cơ quan bị ảnh hưởng như tim, gan, thận và đường tiêu hóa.
Từ “amylon” được sử dụng lần đầu tiên vào năm 1834 bởi nhà thực vật học người Đức Matthias Schleiden để mô tả tinh bột sáp trong thực vật. Sau đó, Rudolph Virchow đã đặt ra từ “amyloid” vào năm 1854 để mô tả tình trạng các mô lắng đọng như cellulose khi tiếp xúc với iốt. Các nhà nghiên cứu bệnh học đã phát hiện ra rằng amyloid dưới ánh sáng bình thường có màu hồng và chuyển sang màu xanh dưới ánh sáng phân cực.
Phân loại Amyloidosis
Có nhiều cách phân loại các thể Amyloidosis.
- Theo vị trí: Thể toàn thân và Thể khu trú
Thể toàn thân, hay còn gọi là thể hệ thống: Bệnh có biểu hiện ở nhiều cơ quan làm tình trạng bệnh trầm trọng hơn và nguy cơ tử vong cao. Trong thể này, người ta chia thành Amyloidosis nguyên phát, thứ phát và di truyền.
Thể khu trú: Bệnh khu trú ở một cơ quan với sự lắng đọng có giới hạn, do vậy, thường chỉ ảnh hướng đến một cơ quan nhất định. Thể này có tiên lượng bệnh tốt hơn. Amyloidosis cơ quan biểu hiện trên một cơ quan thường là Da, Não, Nội tiết.
- Theo nguyên nhân bệnh sinh: Amyloidosis nguyên phát và Amyloidosis thứ phát
- Trong lâm sàng, bệnh thường được chia dựa trên hóa sinh, tức là theo loại protein chịu trách nhiệm. Các loại thường gặp được trình bày dưới đây.
Trong đó,các loại thường gặp nhất là:
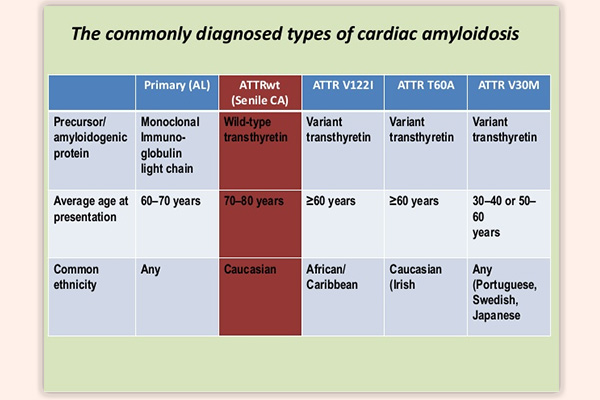
AL (Nguyên phát) Amyloidosis: Là dạng phổ biến nhất của bệnh. Trong thể này, hệ thống miễn dịch của cơ thể tạo ra các dạng kháng thể bất thường gọi là “chuỗi ánh sáng”. Thông thường, các tế bào trong tủy xương được gọi là ” các tế bào plasma, các tế bào này tạo ra các protein kháng thể, giúp cơ thể chống lại nhiễm trùng. Nếu một tế bào plasma không kiểm soát trở thành một tế bào ung thư, nó có thể tạo ra thêm các mảnh kháng thể gọi là “chuỗi ánh sáng” (“L” trong bệnh amyloidosis “AL”). Những chuỗi ánh sáng này lưu thông trong máu, và có thể lắng đọng trong các cơ quan khắp cơ thể, gây tổn thương nội tạng. Thể bệnh AL amyloidosis này có những nét tương đồng lớn với bệnh đa u tủy xương – một bệnh trong đó các dòng vô tính giống hệt của các tế bào sản xuất kháng thể phát triển nhanh chóng, gây nhiều khó khăn trong chẩn đoán và điều trị. Trong đa u tủy, vấn đề chính là sự phát triển của các tế bào bất thường trong tủy xương. Trong bệnh Amyloidosis AL (nguyên phát), vấn đề chính là sự tích tụ các chuỗi ánh sáng được tạo ra bởi các tế bào bất thường. Có thể có sự chồng chéo giữa hai bệnh và bệnh nhân thường được chẩn đoán mắc cả u tủy và bệnh Amyloidosis AL (nguyên phát).
AA (Thứ phát) Amyloidosis: Được đặc trưng bởi một loại protein gọi là “amyloid huyết thanh A”. Protein này được cơ thể sản xuất để đáp ứng với viêm hoặc nhiễm trùng. Hàm lượng protein cao không gây ra sự lắng đọng amyloid trong thời gian ngắn, nhưng có thể dẫn đến sự tích lũy amyloid trong một thời gian dài. AA Amyloidosis có thể do các bệnh dẫn đến tình trạng viêm mạn tính (như viêm khớp dạng thấp được kiểm soát kém) hoặc tình trạng nhiễm trùng mãn tính (như bệnh lao mãn tính). Thể này thường ảnh hưởng đến thận, gan, lá lách.
Familial ATTR Amyloidosis: Biểu hiện hội chứng lâm sàng là bệnh đa thần kinh có tính di truyền. Đây là một bệnh di truyền, trong đó cơ thể tạo ra một dạng đột biến của một protein “transthyretin”. Transthyretin được viết tắt là “TTR” và là lý do căn bệnh này được gọi là bệnh amyloidosis ATTR gia đình. Bệnh amyloidosis ATTR gia đình chủ yếu ảnh hưởng đến tim và dây thần kinh. Mặc dù đây là một bệnh rất nghiêm trọng, tiên lượng của bệnh amyloidosis ATTR gia đình nói chung tốt hơn so với bệnh amyloidosis AL, vì nó thường tiến triển với tốc độ chậm hơn.
Wild-Type (Senile) ATTR Amyloidosis: Bệnh amyloidosis ATTR kiểu hoang dã (tương tự như bệnh amyloidosis ATTR gia đình, nhưng protein lắng đọng là protein transthyretin bình thường, không bị đột biến. Bệnh thường khởi phát từ 65 tuổi trở lên. Bởi vì amyloid tích lũy chậm trong dạng bệnh này, tiên lượng thường tốt hơn so với amyloidosis AL (nguyên phát) và amyloidosis ATTR gia đình.
Sinh bệnh học
Protein fibril amyloid là một protein được lắng đọng dưới dạng fibril không hòa tan, chủ yếu ở các không gian ngoại bào của các cơ quan và mô do kết quả của sự thay đổi trình tự trong quá trình gấp protein dẫn đến tình trạng gọi là amyloidosis. Một protein fibril amyloid xảy ra trong các mô tế bào dưới dạng các sợi cơ cứng, không phân nhánh có đường kính khoảng 10nm.
Triệu chứng lâm sàng, chẩn đoán và điều trị đối với từng thể bệnh
AL Amyloidosis (Amyloidosis nguyên phát)
Loại amyloidosis phổ biến nhất xảy ra trong tập hợp các amyloidosis. Tuy nhiên, đây là một bệnh tương đối hiếm gặp với tỷ lệ mắc 5,1-12,8 người trên một triệu người và chỉ có khoảng hơn 2000 trường hợp mới được chẩn đoán tại Hoa Kỳ hàng năm. Tỷ lệ mắc bệnh ở nam và nữ là 3: 2.
Triệu chứng lâm sàng:
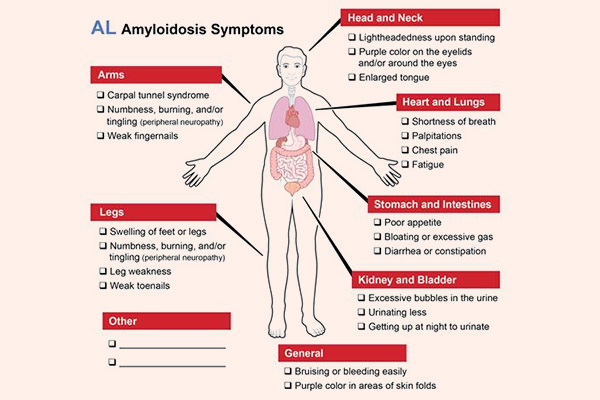
Toàn thân: Gần 90% bệnh nhân sẽ bị mệt mỏi nặng, sụt cân và phù nề. Phù có thể do nhiều nguyên nhân, có thể do hạ đường huyết (do liên quan đến thận, ruột hoặc gan), suy tim phải hoặc mạch máu không chịu được áp lực thủy tĩnh của dòng máu dẫn đến thoát nước ra ngoại vi.
Các triệu chứng lâm sàng khác phụ thuộc vào các cơ quan bị ảnh hưởng ở một bệnh nhân nhất định. Ảnh hưởng đến gan được ghi nhận ở 15-25% bệnh nhân, bệnh lý thần kinh ở 15-20% và tim lên tới 50%. Trong đó, suy tim có triệu chứng chiếm 25-33% bệnh nhân, thể này có thời gian sống trung bình khoảng 6 tháng. Trên thận biểu hiện lâm sàng nổi bật như hội chứng thận hư. Khi các kháng thể lắng đọng ở đường tiêu hóa gây ra bệnh lưỡi to (macroglossia), tiêu chảy do kém hấp thu, giảm trương lực dạ dày và táo bón. Tổn thương đa cơ quan (trên 3 cơ quan) được thống kê trong 30% bệnh nhân.
Ngoài ra, Amyloidosis cũng có thể gây hạ huyết áp thế đứng, suy giáp, thâm nhiễm hồng cầu lưới. Amyloid lắng đọng trong phổi, gây tràn dịch màng phổi, lắng đọng ở da, gây nổi hạch và lách to.
Do amyloid thâm nhiễm mạch máu, rối loạn chức năng của amyloid dẫn đến rối loạn tiểu cầu và đặc biệt là yếu tố đông máu X gây ra ban xuất huyết quanh hốc mắt và các biểu hiện khác của chứng loạn sản xuất huyết. Yếu tố X có khuynh hướng liên kết với các sợi cơ amyloid và sự thiếu hụt của nó không thể dễ dàng được khắc phục bằng cách sử dụng huyết tương vì yếu tố truyền X liên kết nhanh chóng với các mảng amyloid lớn trong gan và lá lách mà không gây ra sự gia tăng của yếu tố X trong máu.
Chẩn đoán
Chẩn đoán AL amyloidosis có thể được thực hiện bằng nhiều cách, trong đó, bước quan trọng đầu tiên là xem xét các triệu chứng lâm sàng. Điện di protein huyết thanh với điện di immunofixation (SPEP / IFE) là một xét nghiệm sàng lọc được sử dụng thường xuyên, có thể được kiểm tra được ở khoảng 25% những người mắc bệnh amyloidosis. Khi được sử dụng kết hợp với SPEP / IFE, điện di protein nước tiểu 24 giờ với IFE có thể phát hiện 90% những người bị ảnh hưởng bởi bệnh amyloidosis.
Gần đây, các nhà lâm sàng đưa thêm xét nghiệm chuỗi ánh sáng tự do trong huyết thanh, định lượng số lượng và loại thậm chí một lượng rất nhỏ chuỗi ánh sáng tự do.
Sinh thiết vẫn là tiêu chuẩn vàng trong xét nghiệm xác định Amyloidosis. Nếu các xét nghiệm trước đó không đủ độ tin cậy, sinh thiết của các cơ quan bị ảnh hưởng có thể được xem xét, nhưng các rủi ro cần được cân nhắc cẩn thận theo xu hướng bệnh nhân amyloidosis bị biến chứng xuất huyết.
Các xét nghiệm không xâm lấn vẫn được ưu tiên hơn.
Điều trị:
Bệnh nhân mắc bệnh amyloid AL không chỉ có khối u ác tính về huyết học, mà còn xuất hiện rối loạn chức năng tiến triển của một hoặc nhiều cơ quan. Một xử trí nhanh là rất cần thiết để bắt giữ các tổn thương nội tạng tiến triển và có thể giải cứu chức năng của nó. Ngoài ra, sự tham gia của multygan amyloid làm cho những bệnh nhân này đặc biệt dễ bị ảnh hưởng bởi hóa trị.
Việc điều trị bệnh amyloidosis AL đã phát triển và có những bước tiến nhanh chóng trong những năm gần đây, phản ánh những thay đổi trong điều trị đa u tủy. Năm 1978, Robert Kyle đã báo cáo một thử nghiệm mù đôi, ngẫu nhiên, có đối chứng giả dược đánh giá hiệu quả của Melphalan cộng với thuốc Prednisolon (liệu pháp được lựa chọn tại thời điểm đó đối với u tủy) trong điều trị bệnh amyloidosis. Ông phát hiện ra rằng một nửa số bệnh nhân được điều trị bằng Melphalan và Prednison đã cải thiện và một phần tư vẫn ổn định, nhưng tỷ lệ sống sót chung không cải thiện. Nghiên cứu kể từ đó cho thấy tỷ lệ đáp ứng tổng thể đối với Melphalan kết hợp với thuốc Prednison là khoảng 30% và xảy ra chậm. Thời gian sống sót trung bình từ 12-17 tháng nhưng được kéo dài ở những bệnh nhân đáp ứng, trong đó 78% sẽ sống sót sau 5 năm. Chỉ riêng Dexamethasone đã đạt được tỷ lệ đáp ứng chung là 45% (đáp ứng hoàn toàn 24%), trong khi Melphalan cộng với Dexamethasone đạt được tỷ lệ đáp ứng chung là 51-68%. Thời gian sống trung bình với Dexamethasone đơn độc là 31 tháng, 70 tháng với phác đồ Melphalan cộng với Dexamethasone trừ khi bệnh nhân bị amyloidosis tim nặng, trong trường hợp sống sót giảm xuống còn khoảng 24 tháng. Tuy tỷ lệ sống và thời gian sống thay đổi không nhiều nhưng sự cải thiện về triệu chứng lâm sàng, cụ thể là đáp ứng huyết học (giảm đáng kể hoặc ức chế hoàn toàn sản xuất paraprotein) chỉ chuyển thành phản ứng của cơ quan (chỉ số cải thiện chức năng cơ quan) khoảng một nửa thời gian và với tốc độ chậm hơn.
Ở những bệnh nhân có nguy cơ thấp (dưới 65 tuổi, với ≤2 cơ quan chính có liên quan, NT-proBNP <332 ng / L, troponin tim T <0,035 g / L hoặc troponin tim I <0,1 μg / L, độ thanh thải creatinine ≥50 mL / phút, khả năng khuếch tán phổi 50% và huyết áp tâm thu> 90 mmHg) SCT với Melphalan 200 mg / m 2 có thể được coi là liệu pháp đầu tay.
Bệnh nhân có nguy cơ cao, mắc bệnh amyloidosis tim tiến triển, và tăng nhiệt độ (cTnI> 0,1 μg / L hoặc cTnT> 0,035 g / L) và NT-proBNP (> 332 ng / L) và Hiệp hội Tim mạch New York (NYHA) hoặc tình trạng hiệu suất IV hoặc ECOG 3 (không phải do bệnh đa dây thần kinh), có thời gian sống trung bình ngắn (3,5 tháng) cần một liệu pháp tối ưu hơn.
Trong vài năm gần đây, các loại thuốc điều hòa miễn dịch (IMiD) như Thalidomide và Lenalidomide và chất ức chế proteasome Bortezomib đã thay đổi đáng kể phạm vi điều trị bệnh đa u tủy và chúng ngày càng được áp dụng cho bệnh amyloidosis. Thalidomide, IMiD đầu tiên được sử dụng lâm sàng, cộng với dexamethasone cho tỷ lệ đáp ứng tổng thể là 48% và tỷ lệ đáp ứng hoàn thành là 19%, và thời gian đáp ứng là ngắn đáng kể ở mức 3,6 tháng. Tuy nhiên, cần thận trọng trong việc sử dụng các loại thuốc này trên từng đối tượng bệnh nhân, vì thalidomide có nguy cơ gây ra bệnh thần kinh và táo bón, các triệu chứng có thể gây rắc rối cho bệnh nhân amyloidosis ngay cả trước khi điều trị. Lenalidomide, IMiD thứ hai cộng với dexamethasone cho tỷ lệ đáp ứng tổng thể là 41-67% với tỷ lệ đáp ứng hoàn toàn là 29%. Thời gian đáp ứng huyết học là 6,2 tháng và phản ứng nội tạng có thể xảy ra ở mức trung bình 9,4 tháng. Lenalidomide ít gây ra bệnh lý thần kinh nhưng có thể gây ứ nước và gây ức chế tủy đáng kể ở hơn một nửa số bệnh nhân. Cả thalidomide và lenalidomide đều có thể gây ra các biến cố huyết khối, do đó cần phải điều trị dự phòng bằng aspirin ở mức liều tối thiểu. Riêng Bortezomib cho tỷ lệ đáp ứng tổng thể là 50%, với tỷ lệ đáp ứng hoàn toàn là 20% và thời gian đáp ứng huyết học là 1,2 tháng thậm chí còn ngắn hơn. Thậm chí có thể có đáp ứng tốt hơn khi bổ sung dexamethasone vào bortezomib, với tỷ lệ đáp ứng tổng thể là 54-80% và tỷ lệ đáp ứng hoàn thành là 15-31%.
Trong những người được đánh giá đủ điều kiện, phương pháp điều trị amyloidosis hiệu quả nhất có thể là ghép tế bào gốc tạo máu tự thân cho dù có được áp dụng hóa trị liệu trước đó hay không. Các bệnh nhân có rối loạn chức năng cơ quan ngay từ ban đầu không được khuyến cáo sử dụng liệu pháp này.
Điều trị bổ trợ cho bệnh nhân amyloidosis bao gồm: Hạn chế muối và sử dụng hợp lý thuốc lợi tiểu. Bởi vì chức năng tim trong bệnh amyloidosis thường phụ thuộc vào tiền gánh (thể tích tuần hoàn), điều quan trọng là tránh giảm quá mức thể tích nội mạch của bệnh nhân. Thuốc chẹn kênh canxi có thể gây che lấp các triệu chứng lâm sàng do tác dụng kích thích âm tính của chúng, và thuốc ức chế men chuyển angiotensin và thuốc ức chế thụ thể angiotensin thường được dung nạp kém do xu hướng gây hạ huyết áp. Digoxin cũng nên được sử dụng một cách thận trọng vì nó có thể liên kết với các sợi cơ amyloid và gây độc tính tại tim ngay cả trong môi trường nồng độ digoxin trong huyết thanh nằm trong khoảng điều trị. Fludrocortisone là một lựa chọn ít được lựa chọn hơn do xu hướng gây ứ nước.
AA Amyloidosis (thứ phát)
Bệnh amyloidosis (hay trước đây được gọi là amyloidosis thứ phát [AA]) là một rối loạn đặc trưng bởi sự lắng đọng của các sợi fibril ở mô ngoại bào bao gồm các mảnh và / hoặc amyloid huyết thanh nguyên vẹn protein A (SAA). Trong khi amyloidosis chuỗi nhẹ (AL) thường gặp ở các nước phát triển, amyloid A (AA) phổ biến hơn ở các nước đang phát triển. Phổ của bệnh amyloidosis AA đã thay đổi trong những thập kỷ gần đây do: sự gia tăng tuổi trung bình trong chẩn đoán; sự gia tăng phần trăm về tần suất bệnh amyloid AL nguyên phát liên quan đến loại AA; và một sự thay đổi đáng kể trong dịch tễ học của các bệnh tiềm ẩn.
AA amyloidosis có thể làm trầm trọng thêm các tình trạng viêm mãn tính, bao gồm viêm khớp dạng thấp (RA), viêm khớp tự phát ở trẻ vị thành niên, viêm cột sống dính khớp (AS), bệnh viêm ruột, nhiễm trùng mãn tính.
Triệu chứng lâm sàng

Bệnh Amyloidosis chủ yếu ảnh hưởng lên thận và các cơ quan ngoài thận khác là gan và lá lách.
Thuật ngữ hội chứng thận hư đề cập đến một nhóm các triệu chứng báo hiệu các vấn đề nghiêm trọng về thận. Triệu chứng đầu tiên của bệnh amyloidosis thường là xuất hiện protein trong nước tiểu. Thông thường protein niệu (protein trong nước tiểu) sẽ tăng rất nhanh và tổn thương thận càng trầm trọng hơn. Bệnh nhân mất quá nhiều protein (albumin) trong máu qua nước tiểu, dẫn đến tình trạng sưng mắt cá chân và chân (phù lớn, trắng, mềm, ấn lõm). Mức cholesterol cao cũng là một phần của hội chứng này do cơ thể tăng tổng hợp để cải thiện áp suất keo của máu. Do đó, bệnh nhân có thể bị suy thận và cần phải lọc máu.
Bệnh amyloidosis liên quan đến các cơ quan khác ngoài thận. Bệnh nhân có thể gặp phải triệu chứng gan, lách phình to. Bệnh lý thần kinh thực vật thường gặp với các triệu chứng hạ huyết áp thế đứng (huyết áp thấp khi đứng), mất trương lực đường tiêu hóa (chậm làm rỗng dạ dày) và tiêu chảy hoặc táo bón. Sự tích lũy amyloid trong tim gây suy tim sung huyết và rối loạn nhịp tim (nhịp tim không đều) có thể phát triển trong quá trình sau của bệnh, tuy nhiên, biến cố trên tim là khá hiếm.
Các triệu chứng ở bệnh nhân mắc bệnh amyloidosis có thể bị hiểu nhầm là các triệu chứng liên quan đến nhiễm trùng mãn tính hoặc viêm. Lúc đầu, người bệnh có thể có các triệu chứng như sụt cân, yếu và sưng (phù). Lý do được đưa ra là vì các rối loạn tiên phát của bệnh nhân cũng có thể gây ra những vấn đề này.
Trong một tình huống ngược lại, bệnh amyloidosis có thể được tìm thấy trước tiên, trước khi một bệnh hoặc tình trạng khác được xác định. Ví dụ, bệnh amyloidosis có thể được chẩn đoán là kết quả của hội chứng thận hư và sau đó có thể dẫn đến việc điều tra một tình trạng tiềm ẩn không được chẩn đoán. Vì vậy, mặc dù amyloidosis AA được kích hoạt bởi một rối loạn tiên phát, không phải lúc nào cũng có nghĩa là rối loạn tiên phát đã được phát hiện và chẩn đoán trước đó.
Chẩn đoán
Chẩn đoán được xác nhận dựa trên sự tham gia của các cơ quan lâm sàng và chứng minh mô học của tiền gửi amyloid. Chẩn đoán không thể được xác nhận dựa trên việc tìm thấy tiền gửi amyloid trong sinh thiết gián tiếp trong trường hợp không có sự tham gia của các cơ quan lâm sàng hoặc trong tình trạng có khuynh hướng, ngay cả với các protein amyloidogen tăng cao trong huyết thanh và không có bằng chứng mô học về tổn thương nội tạng. 3 Sau khi chẩn đoán ban đầu được thực hiện, phân nhóm amyloid phải được xác định và đánh giá sự tham gia của cơ quan hệ thống. Do không có tiêu chí tham chiếu cụ thể để xác định tổn thương nội tạng trong bệnh amyloidosis, nên những người được chấp nhận cho loại AL thường có thể được áp dụng, mặc dù thận trọng
Lựa chọn vị trí sinh thiết:
- Chọc hút mô mỡ dưới da: Thủ thuật đơn giản này có thể được thực hiện trong môi trường ngoại trú bằng cách hút hai đến năm mẫu mỡ bụng bằng kim G được nối với ống tiêm 10 mL. Xét nghiệm này có độ đặc hiệu của chẩn đoán amyloidosis cao (93% đến 100%), độ nhạy thay đổi trong khoảng 57% đến 82%. Do đó, thủ thuật này được các chuyên gia khá ưa thích.
- Sinh thiết niêm mạc trực tràng: Nhiều nghiên cứu hỗ trợ sử dụng sinh thiết niêm mạc trực tràng và dưới niêm mạc, với độ nhạy khoảng 75% đến 85% để phát hiện sự lắng đọng amyloid. Tuy nhiên, một số loạt nghiên cứu đã cho thấy độ nhạy thấp hơn so với hút mỡ bụng và độ xâm lấn phức tạp hơn.
- Sinh thiết tuyến nước bọt nhỏ: Kỹ thuật này cung cấp độ nhạy từ 83% đến 100% để chẩn đoán cả amyloidosis AL và bệnh amyloidosis. Nó cũng hữu ích trong việc phát hiện transthyretin bị đột biến, mặc dù nó có độ nhạy thấp hơn trong bệnh amyloidosis liên quan đến 2 -microglobulin. Sinh thiết của niêm mạc nướu cũng có thể có ý nghĩa trong chẩn đoán, nhưng có độ nhạy thấp hơn.
- Các cơ quan nhạy cảm sinh thiết khác bao gồm da, lưỡi, dây thần kinh ngoại biên, nội tâm mạc và tủy xương, có độ nhạy khác nhau, tùy thuộc vào loại amyloidosis và mức độ tham gia hệ thống.
Điều trị
Do sự đa dạng của nguyên nhân gây bệnh amyloidosis, không có chiến lược điều trị chung cho bệnh amyloidosis. Do đó, tất cả các lựa chọn điều trị nên dựa trên sự kiểm soát tốt hơn căn bệnh tiềm ẩn, điều trị hỗ trợ cơ quan đầy đủ và giảm triệu chứng.
- Điều trị bệnh lí nền
Kiểm soát các bệnh lý tiềm ẩn có tác dụng làm giảm nồng độ các chất phản ứng trong giai đoạn cấp tính tiếp theo bao gồm nồng độ SAA huyết thanh, là chiến lược hiệu quả nhất để ổn định hoặc thậm chí giảm thiểu lắng đọng amyloid. Phẫu thuật cắt bỏ khối u gây viêm cho phép giảm đáng kể nồng độ chất phản ứng giai đoạn cấp trong huyết thanh và trong lắng đọng amyloid được phát hiện bởi quang phổ trong nhiều trường hợp. Sự thuyên giảm của hội chứng thận hư liên quan đến amyloidosis cũng đã đạt được khi điều trị bệnh lao ở bệnh nhân lao. Colchicine liều cao (1,5 – 2 mg / ngày) có hiệu quả trong việc kiểm soát viêm toàn thân trong các hội chứng viêm tự phát và có thể giảm bệnh amyloidosis liên quan. Hơn nữa, các kháng thể kháng IL-1, như Anakinra hoặc Canakinumab, đã được chứng minh là có hiệu quả như các liệu pháp đầu tiên trong một số hội chứng viêm tự phát, cũng có thể chứng minh hiệu quả ở những bệnh nhân mắc FMF cấp tính. Bên cạnh đó, thuốc điều hòa miễn dịch cũng đã được chứng minh là cực kỳ hữu ích trong việc kiểm soát sự tiến triển của protein niệu do amyloidosis và cải thiện sự duy trì kiểm soát trong một số bệnh viêm khớp và bệnh viêm ruột. Một số đại diện của nhóm điều biến miễn dịch như: Chlorambucil, cyclophosphamide, tacrolimus và kháng thể kháng TNF-alpha hoặc kháng IL-6, như Infliximab, etanercept hoặc tocilizumab.
- Điều trị nhắm mục tiêu lắng đọng amyloid
Hiểu rõ về các cơ chế sinh lý bệnh cơ bản của bệnh là sự lắng đọng amyloid đã cho phép phát triển các chiến lược điều trị mới, đặc biệt là các mục tiêu hình thành các protein amyloid. Theo cơ chế này, tocilizumab, một kháng thể đơn dòng kháng IL-6 được tổng hợp, đã cho hiệu quả đáng trông đợi trong việc làm giảm mức độ SAA trong tuần hoàn và kiểm soát sự tiến triển của bệnh amyloidosis trong một số bệnh khớp tự miễn. Tác dụng của nó là độc lập với bệnh lý nền và không bị ảnh hưởng bởi các chất điều hòa miễn dịch đã nói ở trên hoặc với abatacept và rituximab. Tocilizumab không chỉ được thử nghiệm thành công trong bệnh viêm khớp dạng thấp hoặc viêm khớp mãn tính ở vị thành niên, mà còn trong bệnh lao.
Dimethyl sulfoxide là một dẫn xuất của lipoprotein nội bào. Nó đã được thử nghiệm ở những bệnh nhân bị amyloidosis đường tiêu hóa và thận và cho kết quả có thể làm giảm mức độ phản ứng giai đoạn cấp tính và cải thiện feedback ngược của đường tiêu hóa trong khi giảm lắng đọng amyloid cục bộ. Tuy nhiên, chức năng thận được cải thiện chỉ đạt được một cách hữu hình ở những bệnh nhân bị protein niệu nhẹ khi bắt đầu điều trị.
Eprodisate là một chất có trọng lượng phân tử thấp tương tự như heparan sulfate. Bằng cách liên kết cạnh tranh với các vị trí liên kết GAG, nó ức chế sự trùng hợp của các sợi cơ amyloid và ngăn chặn sự tích lũy của amyloid. Các thử nghiệm pha II (thử nghiệm trên người khỏe mạnh quy mô nhỏ) cho thấy sự ổn định chức năng thận trong 42% trường hợp, mặc dù thuốc không thể thay đổi nồng độ SAA huyết thanh và không có tác dụng đáng kể đối với protein niệu và không cải thiện tỷ lệ sống sót chung.
Heparin và statin cũng có tác dụng có lợi đối với kết quả của bệnh amyloidosis AA. Heparin có thể làm chậm tiến trình phát triển của bệnh bằng cách phá vỡ các liên kết ổn định giữa GAG và SAA trong các vị trí lắng đọng, theo cách tương tự như của eprodisate; Statin dường như phát huy tác dụng của chúng thông qua sự ức chế con đường isoprenoid bằng cách đặc biệt ngăn chặn farnesyltransferase. Cơ chế này được biểu hiện mạnh mẽ ở một số bệnh viêm tự miễn, chẳng hạn như tăng hồng cầu miễn dịch D với hội chứng sốt tái phát, trong đó amyloidosis rất hiếm mặc dù viêm nhiễm mạnh, tái phát.
- Điều trị hỗ trợ
Mặc dù các triệu chứng của chứng rối loạn thần kinh thực vật rất hiếm gặp ở bệnh amyloidosis, hạ huyết áp thế đứng nghiêm trọng có thể dẫn đến ngất tái phát. Trong những trường hợp này, fludrocortison hoặc midodrine có thể hữu ích. Hơn nữa, bệnh tiêu chảy hoặc bệnh lý gây mất mát protein có thể được cải thiện bằng cách sử dụng kháng sinh để làm giảm sự phát triển quá mức của vi khuẩn, prednison hoặc sự kết hợp của thuốc prednisolon và octreotide cũng có thể là các lựa chọn tiềm năng. Trong trường hợp tổn thương nội tạng tiến triển, việc cấy ghép có thể được xem xét, đặc biệt là nếu bệnh viêm nhiễm tiềm ẩn đã được kiểm soát.
- Một số liệu pháp điều trị mới
Hầu hết các mục tiêu nghiên cứu mới tập trung vào việc kiểm soát và giảm lắng đọng amyloid trong mô. Kháng thể chống SAP mới R-1- [6- [R-2-carboxy-pyrrolidin-1-yl] -6-oxo-hexanoyl] pyrrolidine-2-carboxylic acid (CPHPC) được mong đợi sẽ cải thiện sự thanh thải của huyết thanh SAP. Trong thử nghiệm trên 31 bệnh nhân bị amyloidosis toàn thân (bất kỳ loại nào), kháng thể này đã đạt được một đáp ứng một phần trong cải thiện chức năng thận, không có tác dụng phụ đáng kể. Trong một nghiên cứu khác, Kluve-Beckerman và cộng sự đã sử dụng các mô hình murine để chặn RNA thông tin phiên mã SAA với hai oligonucleotide bổ sung antisense. Họ đã đạt được mức giảm> 50% mức SAA trong tuần hoàn, cũng như gánh nặng amyloid mô thấp hơn đáng kể. Cuối cùng, các mô hình murine cũng đã được sử dụng để khám phá việc sử dụng clodronate trong sự suy giảm thực bào, cũng có thể đóng vai trò là mục tiêu tiềm năng để ngăn ngừa và điều trị bệnh amyloidosis. Chúng ta có thể mong đợi rằng sẽ có những phương pháp điều trị tốt hơn trong tương lai gần.
Familial ATTR Amyloidosis
Transthyretin amyloidosis gia đình Là một trường hợp hiếm gặp. Các triệu chứng thường khởi phát ở tuổi trưởng thành và tiến triển nặng hơn theo thời gian. Amyloidosis ATTR gia đình là do thay đổi (đột biến) trong gen quy định protein TTR. Gen di truyền là tính trạng trội, nhưng không phải tất cả những người bị đột biến gen TTR sẽ phát triển ATTR amyloidosis.
Triệu chứng
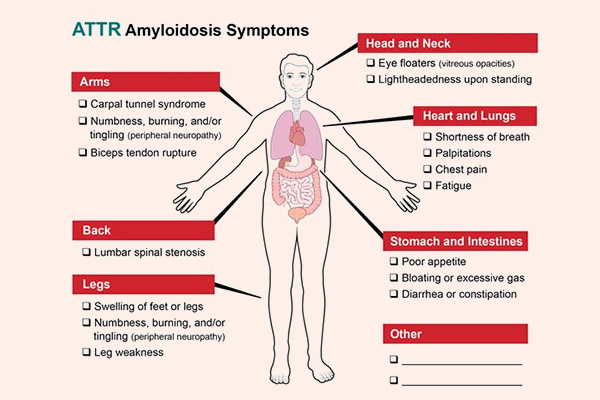
Transthyretin amyloidosis gia đình (FTA) là một tình trạng khởi phát chậm, trưởng thành. Các triệu chứng phụ thuộc vào bộ phận cơ thể bị ảnh hưởng chủ yếu. Các triệu chứng bắt đầu ở các độ tuổi cũng có thể thay đổi theo quốc gia. Ở Bồ Đào Nha và Nhật Bản, những người mắc Transthyretin amyloidosis gia đình thường bắt đầu phát triển các triệu chứng vào cuối những năm 20 đến 40 tuổi. Ở những nơi khác trên thế giới, những người mắc ATTR amyloidosis gia đình có thể không có triệu chứng cho đến sau tuổi 50.
Hệ thần kinh ngoại biên là cơ quan bị ảnh hưởng phổ biến nhất trong ATTR amyloidosis gia đình. Các dạng Transthyretin amyloidosis gia đình khác có thể ảnh hưởng đến não, tủy sống, tim và mắt.
Các triệu chứng của Transthyretin amyloidosis gia đình bao gồm:
- Yếu, tê hoặc đau ở chân và bàn chân dưới
- Hội chứng ống cổ tay (hội chứng chèn ép dây thần kinh giữa) ở cả hai cổ tay
- Bất lực trong sinh hoạt tình dục
- Các vấn đề về tiết niệu, protein niệu
- Rối loạn tiêu hóa: Tiêu chảy hoặc táo bón
- Giảm cân không rõ nguyên nhân
- Khô mắt, tăng nhãn áp
- Rối loạn nhịp tim, phì đại tim
- Bị chóng mặt khi chuyển từ ngồi sang đứng (hạ huyết áp thế đứng)
- Khô mắt và miệng
- Các triệu chứng muộn hơn có thể bao gồm: Yếu cơ và cứng cơ, khó phối hợp động tác, đột quỵ, co giật, mất trí nhớ và suy tim xung huyết.
- Thay đổi màu da,mất thính lực, khó thở và thiếu máu có thể xảy ra nhưng với tần suất thấp.
Chẩn đoán
Chẩn đoán ATTR amyloidosis gia đình (FTA) khá khó khăn vì các dấu hiệu và triệu chứng của FTA thường giống như các thể khác phổ biến hơn. Các chuyên gia thường sử dụng kết hợp các dấu hiệu triệu chứng lâm sàng và xét nghiệm di truyền và xét nghiệm đặc hiệu khác. Xét nghiệm như: Sinh thiết của một khu vực bị ảnh hưởng, xét nghiệm di truyền tìm kiếm một đột biến trong gen TTR.
Điều trị
Không có điều trị đặc hiệu cho ATTR amyloidosis gia đình (FTA) để khỏi bệnh hoàn toàn, nhưng có những phương pháp điều trị có thể ngăn ngừa hoặc trì hoãn sự tiến triển của bệnh. Điều trị tùy thuộc vào cơ quan bị ảnh hưởng và tình trạng bệnh.
Điều trị chính là ghép gan. Thủ tục này loại bỏ nguồn amyloid chính khỏi cơ thể, nhưng amyloid vẫn có thể tích tụ trong tim, não và mắt. Các loại thuốc khác ngăn chặn sự hình thành amyloid và có thể cung cấp một giải pháp không xâm lấn thay thế cho ghép gan. Các phương pháp điều trị khác bao gồm ghép tim, thận, đặt máy tạo nhịp tim, thay thế thể dịch mắt và các loại thuốc dược lí.
Ghép gan là “tiêu chuẩn vàng” để điều trị ATTR amyloidosis gia đình vì nó thay thế nguồn amyloid chính. Nó có thể làm chậm hoặc dừng tiến trình của bệnh lý thần kinh ngoại biên, tuy nhiên bệnh thường vẫn tiến triển ở mắt và não. Cấy ghép nên được thực hiện càng sớm càng tốt trước khi có vấn đề nghiêm trọng về thần kinh.
Một số ít loại thuốc đã được nghiên cứu và phát triển với mục đích làm chậm quá trình tích tụ amyloid dọc theo dây thần kinh và ở các bộ phận khác của cơ thể bao gồm: Tafamidis, Diflunial, và gần đây là Inotersen và Patisiran.
Thuốc lợi tiểu với tác dụng thuốc loại bỏ nước và muối dư thừa ra khỏi cơ thể, thường được sử dụng để kiểm soát tình trạng khi người bệnh bị biến chứng suy tim sung huyết do Amyloidosis. Tương tự, các triệu chứng khác của FTA được điều trị khi chúng phát sinh.
Một số thuốc đã được FDA phê chuẩn:
- Patisiran (Tên thương hiệu: Onpattro) – Được sản xuất bởi Alnylam Enterprises, Inc. Chỉ định được FDA phê chuẩn: Bệnh viêm đa dây thần kinh do bệnh amyloid qua trung gian di truyền.
- Inotersen (Tên thương hiệu: Tegsedi) – Được sản xuất bởi Ionis Enterprises, Inc. Chỉ định được FDA phê chuẩn: Bệnh viêm đa dây thần kinh do bệnh amyloid inated di truyền ở người lớn.
Wild-Type (Senile) ATTR Amyloidosis (ATTRwt-CA)
ATTRwt-CA hiện được cho là có tỷ lệ lưu hành thấp nhất trong số các loại amyloidosis toàn thân khác nhau, có khả năng là do tỷ lệ mắc bệnh thấp, vì nó được tìm thấy trong 10% đến 25% trong số tất cả các bệnh nhân mắc suy tim với phân suất tống máu bảo tồn (HFpEF).
Triệu chứng lâm sàng
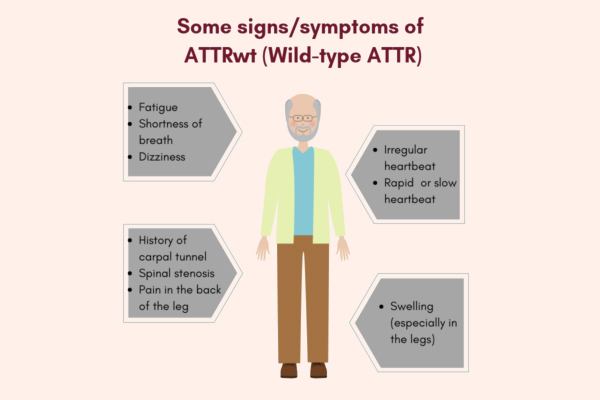
Trái ngược với các dạng amyloidosis khác, ATTRwt-CA hầu như chỉ gây ra bệnh lý rõ ràng về mặt lâm sàng cho tim và các dây thần kinh ngoại biên cũng như các chất lắng đọng không liên quan đến lâm sàng ở gan và phổi. Các biểu hiện về tim của sự lắng đọng protein amyloid không đột biến là chứng phì đại thất trái (LVH) với áp lực tâm thu tăng và giảm phân suất tống máu bảo tồn (HFpEF) điển hình với các triệu chứng khó thở, giảm khả năng lao động, mệt mỏi, đau bụng.
Tuy nhiên, các dấu hiệu và triệu chứng tim này rất giống với các tình trạng bệnh lý khác ảnh hưởng đến bệnh nhân ATTRwt-CA của hầu hết nam giới thường lớn hơn 65 tuổi, chẳng hạn như bệnh động mạch vành, hẹp động mạch chủ, tiểu đường, rối loạn nhịp tim, ngưng thở khi ngủ hoặc béo phì. Bệnh cơ tim phì đại hoặc bệnh tim tăng huyết áp cũng là những triệu chứng gần giống với ATTRwt-CA trên lâm sàng và bằng các phát hiện hình ảnh siêu âm truyền thống, dẫn đến chẩn đoán sai xảy ra khá thường xuyên.
Chẩn đoán
Các yếu tố chính có thể giúp tăng độ nhạy trong chẩn đoán ATTRwt-CA là: Phì đại thất trái mà không có tiền sử suy thận nặng hoặc không kiểm soát được, khởi phát hạ huyết áp, không có khả năng dung nạp thuốc ức chế men chuyển hoặc rối loạn nhịp nhĩ, Rối loạn chức năng nút AV (nhị-thất) cần sử dụng máy tạo nhịp tim vĩnh viễn, Hội chứng ống cổ tay hai bên, Hẹp ống sống. Các triệu chứng suy tim phải cũng liên quan đến ATTRwt-CA với các tĩnh mạch trong tăng cao kéo dài, xung huyết gan, cổ trướng, đầy bụng với cảm giác no sớm và phù chi dưới mạn tính.
Chẩn đoán xác định ATTRwt-CA được thực hiện bằng các biện pháp mô học của sinh thiết tim và kiểu gen. Tuy nhiên, sinh thiết là một thủ tục xâm lấn với các rủi ro tiềm ẩn liên quan và có thể không có sẵn ở nhiều trung tâm do đó, không được sử dụng thường xuyên. Có một số phương thức xét nghiệm không xâm lấn có độ nhạy và độ đặc hiệu khác nhau để chẩn đoán ATTRwt-CA, bao gồm: Điện tâm đồ (EKG), xét nghiệm di truyền, Siêu âm Doppler (Echo), xạ hình, Chụp cộng hưởng từ tim mạch (CMR), cũng như sinh thiết gan và mỡ nếu cần.
Điều trị
Mặc dù có những tiến bộ lớn trong điều trị các thể bệnh Amyloidosis, hiện tại không có liệu pháp chữa trị hoặc điều trị bệnh đặc hiệu đã được chứng minh cho ATTRwt-CA. Lý do có thể khiến ATTRwt-CA không có bất kỳ liệu pháp đã được chứng minh nào: Thứ nhất, đây là một bệnh chưa được công nhận và được chẩn đoán thấp dẫn đến các nhóm bệnh nhân nhỏ để nghiên cứu và hiểu không đầy đủ về sinh lý bệnh và diễn biến của bệnh; thứ hai, bệnh lý này thường xuất hiện ở những bệnh nhân cao tuổi, do đó bị ảnh hưởng bởi tình trạng thể chất và các bệnh lý mắc kèm; thứ ba, quá trình diễn biến của bệnh nhìn chung là chậm do đó hiệu quả điều trị bệnh hoặc điều chỉnh bệnh rất khó đo lường.
Do đó, hầu hết các cân nhắc điều trị cho ATTRwt-CA đều dựa trên ý kiến chuyên gia và quan sát từ tác dụng điều trị đối với loại amyloidosis khác ảnh hưởng đến tim.
- Giảm triệu chứng và chăm sóc hỗ trợ
Nền tảng chính của trị liệu là chăm sóc hỗ trợ nhằm mục đích giảm triệu chứng. Các triệu chứng chính của bệnh nhân mắc ATTRwt-CA là: Nghẹt mũi, mệt mỏi, đau dây thần kinh ngoại biên và hạ huyết áp. Giảm các triệu chứng xung huyết và mệt mỏi liên quan có thể được thực hiện bằng cách sử dụng thuốc lợi tiểu, bao gồm thuốc lợi tiểu quai và thiazide kết hợp với chất đối kháng thụ thể mineralocorticoid để giúp tái hấp thu kali.
Các loại thuốc khác thường được sử dụng trong bệnh cơ tim, chẳng hạn như thuốc chẹn kêch Calci, thuốc ức chế thụ thể ACE-i và angiotensin (ARB), dường như không làm thay đổi tiến triển bệnh và thường dẫn đến tình trạng mệt mỏi và hạ huyết áp.
Thuốc chẹn B thường không được dung nạp vì chúng có thể làm giảm tình trạng suy tim dẫn đến giảm thể tích tuần hoàn có thể dẫn đến đột quỵ và gây ra tình trạng giảm nhịp tim để tăng cung lượng tim.
Thuốc ACE-i và ARB thường làm giảm huyết áp ở bệnh nhân ATTRwt-CA do bệnh lý thần kinh ngoại biên có sẵn ảnh hưởng đến hệ thống thần kinh thực vật của những bệnh nhân mắc phải bệnh lý này.
- Rối loạn nhịp nhĩ
Các triệu chứng liên quan đến ATTRwt-CA trở nên trầm trọng hơn nếu bệnh nhân có tình trạng rối loạn nhịp nhĩ. Việc kiểm soát tốc độ tiến triển rất khó khăn ở những bệnh nhân này vì các thuốc điều trị rối loạn nhịp nhĩ có ảnh hưởng tiêu cực lên bệnh như: Thuốc chẹn kênh beta có thể làm giảm huyết áp ở liều cao, thuốc chẹn kênh canxi bị chống chỉ định khi chúng liên kết với các sợi cơ amyloid gây ra tình trạng xấu đi và digoxin có thể gây độc cho tim. Thuốc chống loạn nhịp tim, chẳng hạn như amiodarone, có thể được sử dụng để kiểm soát nhịp khi bệnh nhân lớn tuổi và được dung nạp tốt vì thường không liên quan đến những thay đổi huyết động khó chịu.
- Máy tạo nhịp tim vĩnh viễn và cấy máy khử rung tim
Xâm nhập tim với các sợi cơ amyloid có thể làm giảm các bất thường của hệ thống dẫn truyền, như rối loạn chức năng nút nhĩ thất. Phần lớn bệnh nhân mắc ATTRwt-CA cần đến máy tạo nhịp tim. Hướng dẫn của hội tim mạch Hoa Kỳ ACC/AHA đề nghị xem xét từng trường hợp để cấy ghép máy khử rung tim cấy ghép (ICD) ở bệnh nhân để phòng ngừa tiên phát và thứ phát các biến chứng bất lợi về tim mạch.
- Phương pháp điều trị nâng cao: thiết bị hỗ trợ tâm thất trái (LVAD) và ghép tim
Bệnh nhân ATTRwt-CA có các triệu chứng suy tim tiến triển và giảm tuổi thọ, những phát hiện có thể đủ điều kiện để được xem xét các biện pháp hỗ trợ cho suy tim tiến triển như LVAD và / hoặc ghép tim. Bệnh nhân mắc ATTRwt-CA thường không được xem xét để ghép tim do biểu hiện bệnh tiến triển, tuổi tác và bệnh kèm theo.
Tài liệu tham khảo
- Systemic Amyloidoses, Luis M. Blancas-Mejía and Marina Ramirez-Alvarado, HHS Author Manuscripts
- The Amyloidoses: Clinical Features, Diagnosis and Treatment, Kelty R. Baker, M.D.a and Lawrence Rice, M.D.b, Methodist Debakey Cardiovasc
- Current treatment of AL amyloidosis, Giovanni Palladini and Giampaolo Merlini, Haematologica
- Familial transthyretin amyloidosis, https://rarediseases.info.nih.gov/diseases/656/familial-transthyretin-amyloidosis
- Wild-type transthyretin cardiac amyloidosis (ATTRwt-CA), previously known as senile cardiac amyloidosis: clinical presentation, diagnosis, management and emerging therapies, Ilia G. Halatchev, Jingsheng Zheng, and Jiafu Ou, 2018 Mar.




{kind=link}