Đái tháo đường là gì?
Đái tháo đường là một tình trạng bệnh lý nội tiết – chuyển hóa khá phổ biến hiện nay. Bệnh đặc trưng bởi tình trạng đường huyết tăng cao kéo dài, đi kèm với đó là một số triệu chứng và biến chứng có thể nguy hiểm đến tính mạng nếu không được phát hiện và điều trị kịp thời.
Đái tháo đường hiện nay đang nổi lên là một trong những bệnh không lây phổ biến và gây tử vong nhiều nhất trên thế giới. Trước đây, bệnh được cho là chỉ phát triển nhiều ở những nước đã phát triển, những nơi có đời sống kinh tế – xã hội cao. Nhưng hiện nay, nó đã có xu hướng ngược lại, đang ngày càng lan rộng ra ở các nước đang phát triển và kém phát triển, mà Việt Nam cũng nằm trong số đó.
Phổ biến nhất là đái tháo đường type 2 (xảy ra với tỷ lệ 90-95%), thường xảy ra ở người lớn, khi cơ thể kháng insulin hoặc không tạo ra đủ insulin. Trong ba thập kỷ qua, tỷ lệ hiện mắc đái tháo đường type 2 đã tăng lên đáng kể ở các quốc gia thuộc mọi mức thu nhập. Đái tháo đường type 1, từng được gọi là đái tháo đường vị thành niên hoặc đái tháo đường phụ thuộc insulin, là một tình trạng mạn tính trong đó tuyến tụy tự sản xuất ít hoặc không có insulin. Đối với những người mắc đái tháo đường, việc tiếp cận với điều trị phải chăng, bao gồm cả Insulin, là rất quan trọng đối với sự sống còn của họ. Một mục tiêu được thống nhất trên toàn cầu để ngăn chặn sự gia tăng đái tháo đường và béo phì đến năm 2025.
Năm 2017, theo IDF (Liên đoàn Đái tháo đường Thế giới), toàn thế giới có khoảng 425 triệu người mắc đái tháo đường. Trung bình trong quần thể dân số trưởng thành (20-79 tuổi), cứ 11 người thì có 1 người mắc đái tháo đường. Ước tính cứ 2 người trưởng thành (20-79 tuổi) bị đái tháo đường thì có 1 người không được chẩn đoán (khoảng 212 triệu người). Tổng chi phí y tế mà các chính phủ hoặc người dân trên toàn thế giới phải chi ra cho đái tháo đường là 727 tỷ USD, chiếm 12% tổng chi phí dành cho y tế trên toàn thế giới. Một vấn đề đáng quan tâm, đó là ¾ số người được chẩn đoán đái tháo đường nằm trong các quốc gia đang phát triển và kém phát triển. Trong tổng số bệnh nhân bị đái tháo đường đó, hơn 1.1 triệu là trẻ em và trẻ vị thành niên bị đái tháo đường type 1. Ước tính đến năm 2045, sẽ có khoảng 629 triệu người mắc đái tháo đường trên toàn thế giới, tương đương với tỷ lệ khoảng cứ 10 người trưởng thành (20-79 tuổi) thì có 1 người bị đái tháo đường, và khi đó gánh nặng y tế phải chi trả cho những bệnh nhân đái tháo đường này sẽ tăng lên đến 776 tỷ USD.
Đầu những năm 2000, tỷ lệ hiện mắc đái tháo đường trong dân số nước ta là khoảng 2-3%. Đến đầu những năm 2010, tỷ lệ dân số mắc đái tháo đường rơi vào khoảng 5-6%. Và cho đến hiện tại (năm 2020), tỷ lệ hiện mắc đái tháo đường ở nước ta tăng lên đến 7-8%.
Ngoài đái tháo đường type 1 và type 2 như đã được mô tả ở trên, còn một vài kiểu đái tháo đường khác chưa được phân loại vào đó, ví dụ như đái tháo đường thai kỳ, đái tháo đường do thận hay đái tháo đường gây ra do thuốc (như do sử dụng các glucocorticoids).
Insulin là gì và vai trò của insulin
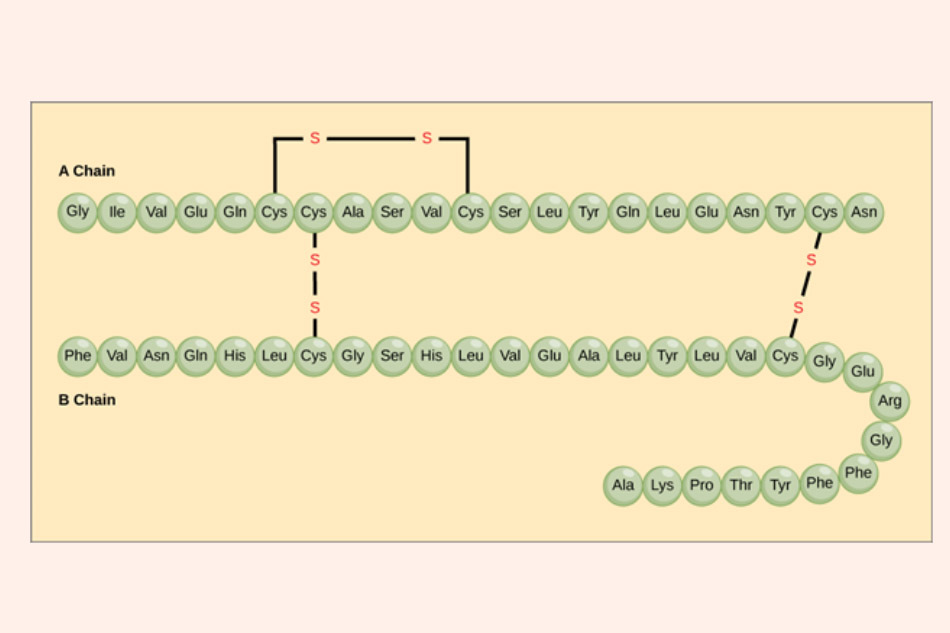
Insulin là một hormone nội tiết do các tế bào β trong tiểu đảo Langerhans của tuyến tụy tiết ra. Về mặt cấu trúc, insulin là một polypeptide có chứa 51 acid amin và chia thành 2 chuỗi: Chuỗi A chứa 21 acid amin, và chuỗi B chứa 30 acid amin. Hai chuỗi được nối với nhau bằng hai cầu disulfide và có thêm một cầu disulfide nằm trong chuỗi A. Phần đặc hiệu đặc trưng cho loài chỉ tập trung vào một số acid amin (bao gồm các acid amin có số thứ tự 8, 9, 10, 11, 12 và 14 của chuỗi A và acid amin thứ 30 của chuỗi B). Các cầu disulfide này đảm bảo cấu trúc không gian của insulin. Nếu chúng bị gãy thì tác dụng của insulin cũng sẽ mất đi.
Ban đầu insulin được tổng hợp dưới dạng preproinsulin. Sau đó nó nhanh chóng được cắt bớt vài acid amin để trở thành dạng dự trữ proinsulin. Proinsulin này khi cần thiết sẽ được cắt tiếp một chuỗi peptide nữa (gọi là C-peptide) để trở thành insulin. Định lượng nồng độ C-peptide trong máu đóng một vai trò quan trọng trong đánh giá khả năng bài tiết insulin của tế bào β đảo tụy.
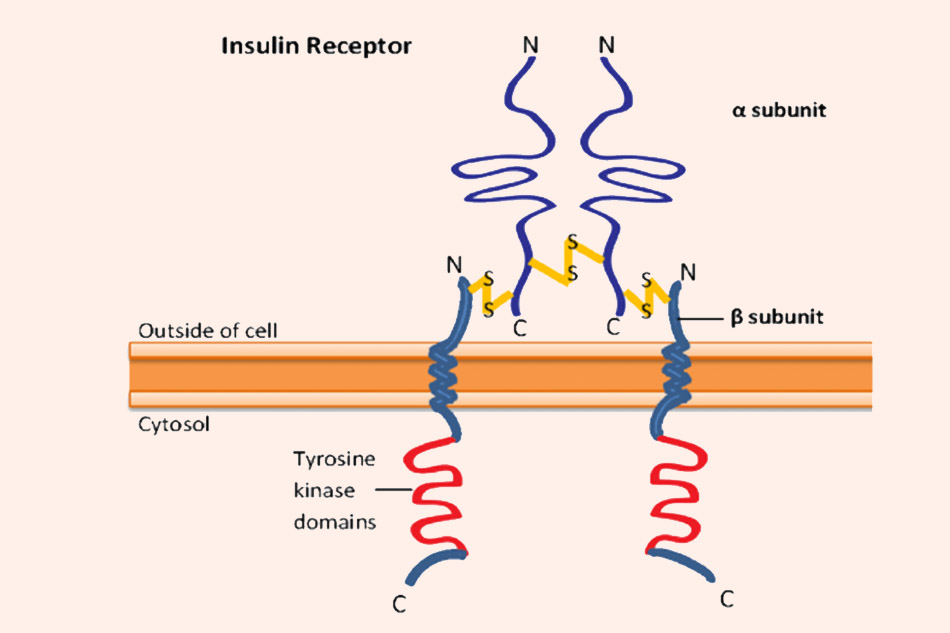
Insulin receptor là một glycoprotein có cấu tạo như sau:
Hai chuỗi α giống nhau, nằm gần nhau trên màng tế bào, có trọng lượng phân tử xấp xỉ 130,000 Da. Hai chuỗi này được nối với nhau bằng cầu nối disulfide và có vị trí đặc hiệu để nhận biết và gắn với insulin.
Hai chuỗi β giống nhau, nằm xuyên màng tế bào, có trọng lượng phân tử cỡ khoảng 90,000 Da. Chuỗi β có hoạt tính tyrosine kinase. Chuỗi β được nối với chuỗi α cũng thông qua cầu nối disulfide.
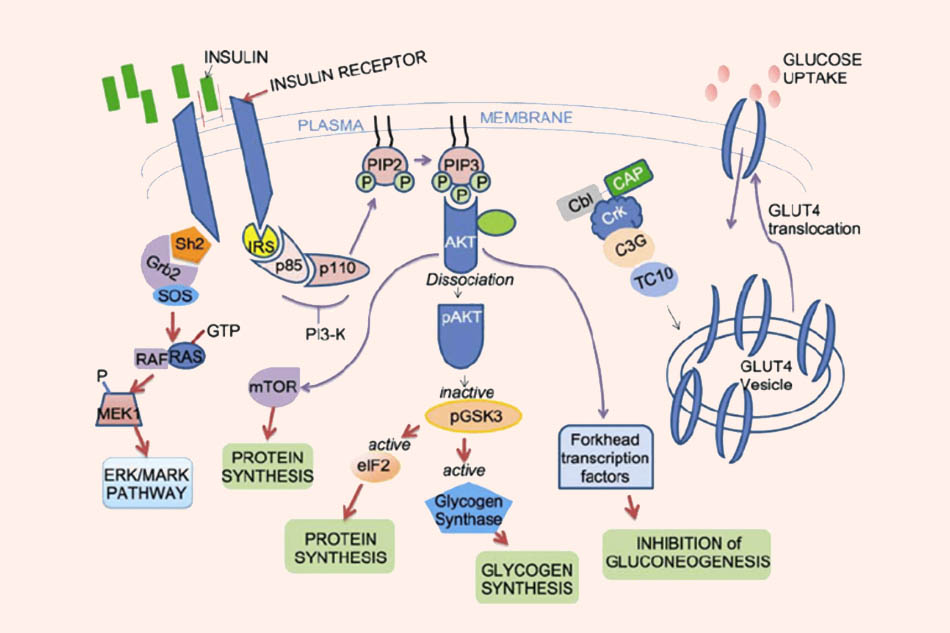
Tác dụng của insulin:
- Chuyển hóa glucid: Insulin làm hạ glucose huyết do tăng vận chuyển glucose vào trong tế bào, đặc biệt là các tế bào gan, cơ vân và mô mỡ. Cơ chế như sau: Khi insulin khi kết hợp với vị trí gắn trên chuỗi α, chuỗi β sẽ bị tác động tự phosphoryl hóa tyrosine của chính bản thân nó. Sau khi gốc tyrosine của chuỗi β bị phosphoryl hóa, các gốc tyrosine của các protein khác trong bào tương cũng lần lượt bị phosphoryl hóa. Điều này tạo ra con đường tín hiệu đến các nang dự trữ trong tế bào. Các nang này chứa những protein vận chuyển glucose, chúng di chuyển tới màng tế bào, hòa màng và hướng chất vận chuyển glucose ra ngoài. Các protein vận chuyển glucose này sẽ tăng cường vận chuyển glucose vào trong tế bào với tốc độ nhanh. Điều này làm hạ glucose máu. Khi nồng độ glucose trong nội bào cao, sự feedback âm xảy ra, insulin rời khỏi vị trí gắn trên chuỗi α, các protein chịu trách nhiệm vận chuyển glucose được thu hồi trở lại trong các nang dự trữ và trở về vị trí ban đầu.
Ngoài ra, insulin cũng tăng tổng hợp và ức chế giáng hóa glycogen tại gan thông quan con đường enzyme, hoạt hóa glycogen synthetase và ức chế glycogen phosphorylase. Insulin cũng ức chế tân tạo đường từ các thành phần khác (acid béo tự do, acid amin).
Hiện nay, các nhà khoa học đã biết tới nhiều hệ vận chuyển glucose. Người ta thường quan tâm đến hai hệ vận chuyển chính là GLUT2 và GLUT4. GLUT4 có nhiều ở tế bào cơ và mô mỡ, có tác dụng hấp thu glucose vào tế bào khi có mặt insulin. Còn GLUT2 có chủ yếu ở tế bào β tiểu đảo, gan, thận, ruột, nó có tác dụng điều hòa giải phóng insulin, từ đó điều hòa nồng độ glucose.
- Chuyển hóa lipid: Tăng tổng hợp và dự trữ, đồng thời giảm thoái hóa lipid. Insulin do đó ức chế sự sinh ra các thể ketone, làm giảm nồng độ acid béo tự do, giảm triglyceride huyết tương.
- Chuyển hóa protid: Insulin làm acid amin tăng vào tế bào, tăng tổng hợp và giảm giáng hóa protein.
Tại sao lại cần phải điều trị đái tháo đường
Cần điều trị đái tháo đường vì bệnh lý này có thể gây ra các biến chứng nguy hiểm, làm giảm chất lượng cuộc sống và tăng tỷ lệ tử vong cho bệnh nhân.
Các biến chứng của đái tháo đường có thể kể đến ở đây là:
Hôn mê do nhiễm toan ketone:
Đây là biến chứng thường gặp trong đái tháo đường type 1. Các tế bào không thể sử dụng glucose làm năng lượng (do thiếu hụt insulin tuyệt đối trong đái tháo đường type 1) nên chúng sử dụng thay thế bằng lipid và protein. Khi lipid bị phá hủy quá nhiều trong thời gian ngắn (đái tháo đường type 1 thường có khởi phát rầm rộ chứ không từ từ như đái tháo đường type 2), chúng tạo ra nhiều thể ketonic trong máu. Các thể ketone này đều có tính acid (ngoại trừ acetone), làm máu bị nhiễm toan, gọi là nhiễm toan ketone (một dạng nhiễm toan chuyển hóa). Nhiễm toan ketone nặng có thể dẫn đến hôn mê và tử vong nếu không được điều trị kịp thời. Trên lâm sàng, có thể dễ dàng chẩn đoán được nhiễm toan ketone thông qua các triệu chứng: nhịp thở Kussmaul (biểu hiện các quá trình liên tục sau trong một chu kỳ thở: hít vào sâu – ngừng thở ngắn – thở ra nhanh – ngừng thở lâu hơn), hơi thở có mùi ketone và nước tiểu cũng có thể ketone.
Hôn mê do tăng áp lực thẩm thấu:
Biến chứng này lại hay gặp trong đái tháo đường type 2 hơn, khi bệnh nhân bị đái tháo đường nặng với đường huyết rất cao. Bệnh nhân hôn mê do tình trạng mất nước nặng do áp lực thẩm thấu tăng cao. Lý do là bởi nồng độ glucose ngoại bào quá cao (do rối loạn tiết insulin và kháng insulin trong đái tháo đường type 2 làm glucose không vào được tế bào) hút nước từ nội bào ra ngoại bào, đồng thời glucose máu cao quá ngưỡng tái hấp thu của thận (cụ thể là tại ống lượn gần, giới hạn tái hấp thu glucose là 10 mmol/L), làm cho glucose thoát ra ngoài theo nước tiểu, làm tăng áp lực thẩm thấu trong lòng ống thận và hút nước ra lòng ống thận, theo đó gây đa niệu thẩm thấu, từ đó mất nước ngoại bào.
Tình trạng hôn mê do tăng áp lực thẩm thấu này không có hoặc ít có thể ketone trong nước tiểu, hơi thở không có mùi của ketone và cũng không có nhịp thở Kussmaul.
Biến chứng mạch máu nhỏ:
- Suy giảm thị lực, mù lòa
Các vi mạch ở mắt bị tổn thương do nồng độ glucose máu cao và quan trọng không kém, do tăng huyết áp (biến chứng mạch máu lớn của đái tháo đường), từ đó làm suy giảm thị lực, đục thủy tinh thể, phù hoàng điểm, bệnh võng mạc đái tháo đường, cuối cùng là dẫn đến mù lòa.
- Bệnh thận mạn, suy thận
Huyết áp cao (biến chứng mạch máu lớn của đái tháo đường) và tổn thương các mao mạch thận, xơ hóa mao mạch cuối cùng dẫn đến giảm mức lọc cầu thận, suy thận mạn. Bệnh thận đái tháo đường diễn biến âm thầm và thường khi được phát hiện thì bệnh đã tiến triển nặng.
- Bệnh lý thần kinh
Các mao mạch cung cấp máu cho các dây thần kinh bị tổn thương cũng vì các lý do tương tự như trên, kết quả là viêm dây thần kinh, hỏng dây thần kinh (gây mất cảm giác vĩnh viễn).
- Nhiễm trùng
Bệnh nhân đái tháo đường thường hay gặp phải nhiễm trùng vì nhiều lý do như: hệ miễn dịch suy yếu do bạch cầu bị hạn chế di chuyển khi nồng độ glucose trong máu cao, các hệ thống vi mạch bị tổn thương làm cho tuần hoàn máu lưu thông đến vùng nhiễm trùng kém, dẫn đến chậm lành hoặc lâu khỏi nhiễm trùng, với các nhiễm trùng ở các cơ quan hay phải tiếp xúc với môi trường ngoài như da, hai nguyên nhân quan trọng có thể góp phần vào nhiễm trùng là thứ nhất, do thị lực suy giảm, bệnh nhân không thể tránh được các va chạm gây ra vết thương hở và cũng khó có thể phát hiện ra chúng, đặc biệt là khi nó ở bàn chân, thứ hai là biến chứng trên thần kinh làm cho bệnh nhân không có cảm giác đau, từ đó không nhận ra và không đề phòng được với nhiễm trùng.
Trường hợp những bệnh nhân bị loét bàn chân đái tháo đường, với ban đầu là loét vô khuẩn, sau đó là loét nhiễm trùng, dẫn đến phải cắt cụt một phần hoặc cả bàn chân không phải là chuyện hiếm.
Biến chứng mạch máu lớn:
- Rối loạn lipid máu
Do glucose không vào được tế bào nên nó không thể được sử dụng để làm năng lượng cho tế bào, thay vào đó, lipid và protein trở thành các nguồn năng lượng chính. Phân giải lipid và protein nhiều gây ra các rối loạn trong thành phần của máu. Bệnh nhân đái tháo đường thường có rối loạn lipid máu như tăng LDL-C (cholesterol lipoprotein tỷ trọng thấp), tăng VLDL-C (cholesterol lipoprotein tỷ trọng rất thấp), tăng triglyceride, tăng cholesterol toàn phần huyết tương cũng như giảm HDL-C (cholesterol lipoprotein tỷ trọng cao). Cả nồng độ LDL-C thấp và HDL-C cao đều là các yếu tố nguy cơ của bệnh tim mạch.
Có nhiều yếu tố có thể gây tăng huyết áp, chẳng hạn như biến chứng vi mạch làm thành mạch xơ cứng, giảm tính đàn hồi, gây tăng sức cản ngoại biến, từ đó gây tăng huyết áp, hoặc có thể do mức lọc cầu thận giảm (cũng là biến chứng vi mạch của đái tháo đường), làm hoạt hóa hệ renin-angiotensin-aldosterone (hệ RAA), gây tăng huyết áp. Tăng huyết áp tự bản thân nó đã có thể làm tăng nguy cơ gặp nhiều biến chứng trên tim, thận, mắt, não và mạch. Trên tim, nó làm tăng nguy cơ đau thắt ngực, nhồi máu cơ tim. Trên thận, nó làm tăng nguy cơ gây suy thận (mối quan hệ giữa tăng huyết áp và bệnh thận mạn tạo thành vòng xoắn bệnh lý). Trên não, tăng huyết áp làm tăng nguy cơ thiếu máu não cục bộ, tai biến mạch máu não. Trên mắt, nó làm tăng nguy cơ tăng nhãn áp, đục thủy tinh thể, suy giảm thị lực, mù lòa (như đã nói ở phần biến chứng mạch máu nhỏ). Trên mạch, tăng huyết áp làm tăng nguy cơ xơ hóa và tổn thương thành mạch, tạo điều kiện cho cholesterol lắng đọng…
- Vữa xơ động mạch, đau thắt ngực và nhồi máu cơ tim
Bệnh mạch vành (bệnh tim thiếu máu cục bộ) là hậu quả của một loạt các biến chứng mạch máu lớn và mạch máu nhỏ ở trên (tăng huyết áp, rối loạn lipid máu, tổn thương vi mạch). Hậu quả của bệnh có thể dẫn đến nhồi máu cơ tim có ST chênh lên (STEMI) hoặc ST không chênh lên (NSTEMI), phải nhập viện chăm sóc tích cực, can thiệp động mạch vành qua da (đặt stent), phẫu thuật bắc cầu chủ – vành… Trường hợp xấu nhất là bệnh nhân có thể tử vong.
Với phụ nữ có thai:
Đái tháo đường thai kỳ làm tăng nguy cơ đẻ non, sẩy thai và nhiễm trùng tiết niệu ở người mẹ. Còn với người con, đái tháo đường thai kỳ gây ra phát triển thai quá mức, hạ glucose máu và các bệnh chuyển hóa ở trẻ sau khi sinh, tăng nguy cơ thai chết lưu, tử vong sau sinh, và hậu quả lâu dài có thể xảy ra là người con sau này cũng có nguy cơ mắc đái tháo đường type 2.
Các nhóm thuốc điều trị đái tháo đường
Các nhóm thuốc hạ đường huyết
Các thuốc trong nhóm này đều có tác dụng hạ đường huyết (hay điều trị triệu chứng) với hiệu lực hạ glucose máu không giống nhau. Chỉ hạ glucose máu giúp giảm các biến chứng vi mạch do đái tháo đường, nhưng không có tác dụng làm giảm các biến chứng đại mạch, hay các biến chứng tim mạch trong đái tháo đường.
Một số ý chính về các thuốc có tác dụng hạ đường huyết được tóm tắt dưới đây.
Insulin:
Insulin sử dụng trong điều trị đái tháo đường không phải là insulin nguyên bản của con người, mà đã được điều chỉnh cấu trúc phân tử cho phù hợp với mục đích điều trị. Ban đầu, Insulin khi mới xuất hiện được chiết tách từ tụy lợn và bò nên rất đắt, đồng thời tác dụng quá nhanh nên phải sử dụng nhiều lần trong ngày, khiến những người dân bình thường khi đó rất khó có thể tiếp cận với việc sử dụng thuốc. Ngày nay, với công nghệ tái tổ hợp (chuyển gen quy định việc tổng hợp insulin vào plasmid của vi khuẩn E.coli), người ta đã có thể tiến hành sản xuất Insulin hàng loạt với giá rẻ, giúp nhiều người dân có thể tiếp cận thuốc hơn, thêm vào đó, các nhà khoa học cũng có thể tùy ý điều chỉnh cấu trúc của phân tử insulin để tạo ra các loại insulin có thời gian tác dụng kéo dài khác nhau như mong muốn.
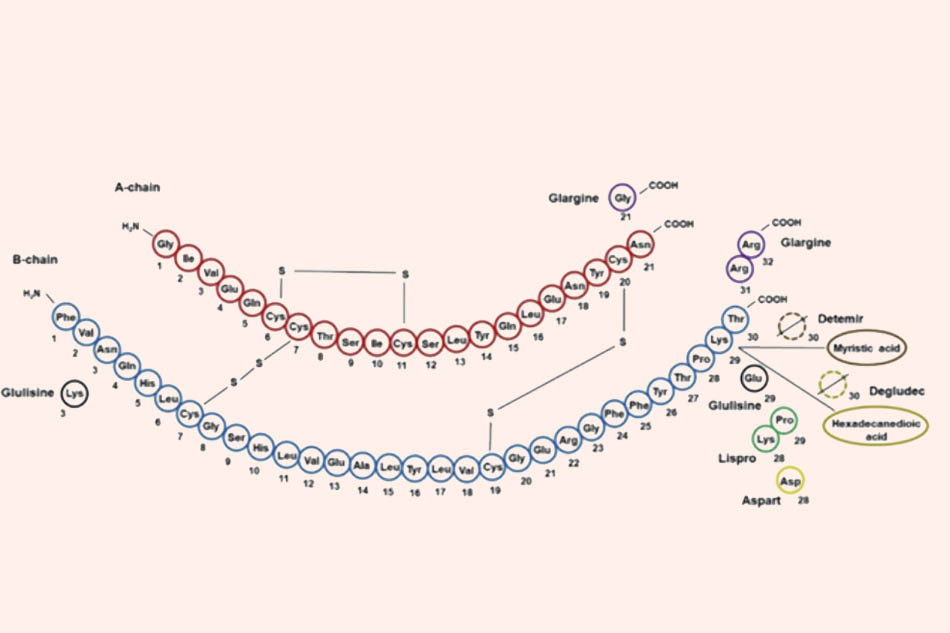
Cấu trúc của các Insulin sử dụng trong điều trị khác với Insulin nội sinh như thế nào?
Insulin được sử dụng theo đường tiêm dưới da (Insulin không thể uống vì sẽ bị acid trong dịch vị phá hủy) là phổ biến nhất và hiện nay có cả dạng Insulin sử dụng theo đường hô hấp. Hiện nay với đường tiêm dưới da, người ta đã làm ra các bút tiêm tự động, đơn giản và dễ sử dụng để giúp cho bệnh nhân có thể tự tiến hành các thao tác mà không cần đến sự trợ giúp của các nhân viên y tế.
Insulin chỉ làm giảm các biến chứng mạch máu nhỏ chứ không làm giảm các biến chứng mạch máu lớn. Hiệu quả làm giảm các biến chứng mạch máu nhỏ tương đương với các thuốc hạ đường huyết đường uống khác, với cả bệnh nhân đái tháo đường type 1 cũng như type 2, mới mắc hay mắc đã lâu.
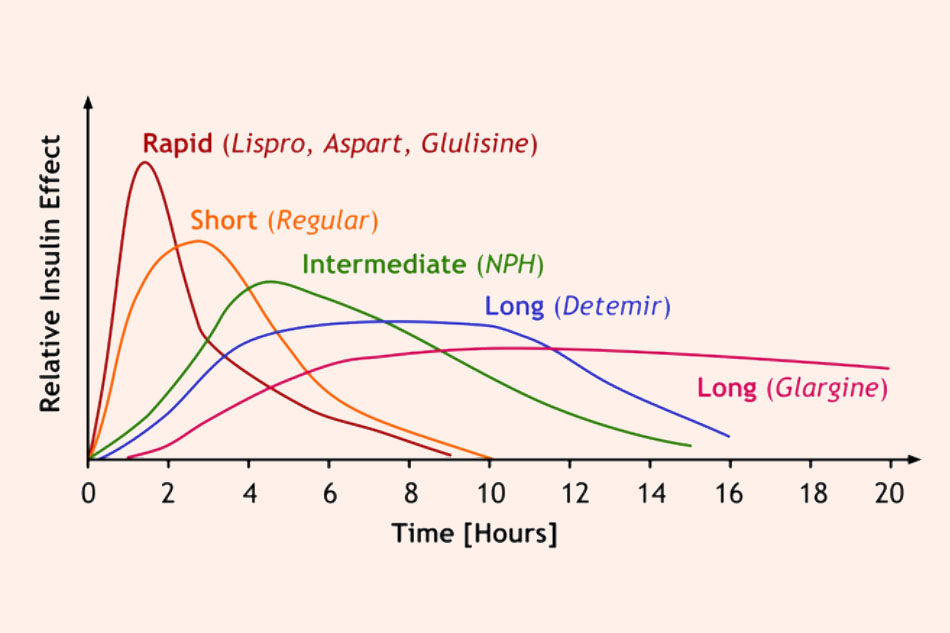
Dựa vào thời gian tác dụng của Insulin, người ta chia các Insulin ra thành các loại:
- Insulin tác dụng nhanh: Insulin lispro, Insulin aspart, Insulin glulisine. Loại này có thời gian khởi phát tác dụng rất nhanh (< 15 phút), nồng độ đỉnh trong huyết tương đạt được chỉ sau 1-2 giờ và thời gian tác dụng cũng không kéo dài (3-5 giờ, với Insulin glulisine thậm chí còn ngắn hơn, chỉ 1-2.5 giờ).
- Insulin tác dụng ngắn (còn gọi là Insulin thường [regular]): Actrapid, Novolin, Velosulin. Loại này khởi phát tác dụng chậm hơn loại trên (30-60 phút), nồng độ đỉnh trong huyết tương đạt được sau 2-5 giờ và thời gian tác dụng cũng dài hơn vài giờ (trừ Velosulin có thời gian tác dụng chỉ 2-3 giờ).
- Insulin tác dụng trung gian: Insulin NPH. Loại này ngoài Insulin còn có chứa protamine và kẽm cùng phenol, không tan ở pH sinh lý (khoảng 7.4). Thời gian khởi phát tác dụng muộn hơn loại trên (1-2 giờ), nồng độ đỉnh trong huyết tương đạt được sau 4-12 giờ và thời gian tác dụng lên đến 18 giờ.
- Insulin tác dụng kéo dài: Insulin detemir, Insulin glargine. Các loại này có tác dụng kéo dài lên đến 24 giờ và chỉ cần một lần tiêm/ngày. Nghiên cứu về dược động học của các thuốc này thường không tìm thấy nồng độ đỉnh trong huyết tương của chúng.
Insulin glargine có điểm đẳng điện pHi = 6.8, do vậy sau khi tiêm dưới da, thuốc nhanh chóng bị kết tủa rồi lại hòa tan trở lại dần dần trở về các dạng monomer và dimer, và chỉ dạng monomer mới có tác dụng. Insulin glargine khởi phát tác dụng sau 1-1.5 giờ, không tìm thấy nồng độ đỉnh (Cpeak) trong huyết tương và thời gian tác dụng lên đến 20-24 giờ.
Phân tử Insulin detemir bám vào albumin ở kẽ nơi tiêm, làm cấu trúc hexamer bền hơn, do đó kéo dài tác dụng của thuốc. Trong máu, Insulin detemir liên kết với albumin huyết tương rồi giải phóng dần dần dưới dạng tự do. Insulin detemir khởi phát tác dụng sau 1-2 giờ, nồng độ đỉnh trong huyết tương đạt được sau 6-8 giờ và tác dụng cũng kéo dài đến 24 giờ.
- Insulin hỗn hợp: Loại này có ưu điểm thời gian khởi phát tác dụng nhanh (như loại Insulin có thời gian tác dụng nhanh và tác dụng ngắn) nhưng cũng đồng thời có tác dụng kéo dài (như loại Insulin có tác dụng kéo dài).
Cơ chế tác dụng: Cơ chế tác dụng của các loại Insulin dược dụng giống như Insulin nội sinh trong cơ thể người. Thuốc có tác dụng hạ glucose huyết ở mọi đối tượng bệnh nhân, và tác dụng phụ thuộc liều.
Chỉ định: Đái tháo đường type 1 (bắt buộc), đái tháo đường type 2 (khi chỉ sử dụng các thuốc khác không đủ để kiểm soát đường huyết), đái tháo đường thai kỳ (bắt buộc).
Tác dụng không mong muốn:
- Dị ứng: Điều này có liên quan mật thiết đến độ tinh khiết của Insulin.
- Hạ kali máu: Điều này được ứng dụng trong điều trị tăng kali máu ở bệnh nhân có bệnh thận cấp hoặc mạn tính. Bệnh nhân sẽ được truyền đồng thời Insulin và glucose.
- Hạ đường huyết quá mức: Hay xảy ra với đái tháo đường type 1 hơn type 2 (vì ở bệnh nhân đái tháo đường type 2, cơ chế feedback âm không còn hiệu quả do cơ thể bệnh nhân không có khả năng tự điều chỉnh bài tiết insulin), điều trị tích cực bằng Insulin (do đưa Insulin nhiều hơn vào thời điểm sau ăn nên dễ hạ glucose huyết hơn).
- Loạn dưỡng mỡ nơi tiêm: Khắc phục tác dụng không mong muốn này bằng cách thay đổi vị trí tiêm liên tục.
- Tăng cân: Do Insulin là hormone tăng đồng hóa. Đây là vấn đề không thể khắc phục được của Insulin. Cần thận trọng và lưu ý về vấn đề này với bệnh nhân thừa cân béo phì (thường gặp trong đái tháo đường type 2).
- Chống chỉ định: Quá mẫn cảm với Insulin hoặc bất cứ thành phần nào của thuốc.
Sulfonylureas:
Các sulfonylureas được khám phá ra lần đầu tiên vào năm 1942. Khi đó, Janbon và các cộng sự đã chú ý thấy rằng một số sulfamides có tác dụng gây hạ đường huyết ở động vật thí nghiệm. Sau đó, một sulfamide là Carbutamide (1-butyl-3-sulfonylurea) đã được tổng hợp và cho thấy tác dụng hạ đường huyết hiệu quả. Nó đã trở thành sulfonylurea đầu tiên được sử dụng để điều trị đái tháo đường trên thị trường, nhưng rất đáng tiếc là sau đó không lâu, nó đã bị rút khỏi thị trường vì tác dụng phụ nặng nề trên tủy xương.
Dù đã ra đời từ rất lâu, nhưng hiện nay các sulfonylureas vẫn là một trong những nhóm thuốc quan trọng nhất trong điều trị đái tháo đường type 2 vì giá thành rẻ và hiệu lực cao.
Các sulfonylureas được chia thành 2 thế hệ với các đại diện:
- Thế hệ 1: Tolbutamide, Chlorpropamide…
- Thế hệ 2: Gliclazide, Glyburide (Glibenclamide), Glimepiride…
Hai thế hệ này không khác nhau về tác dụng hạ glucose huyết khi sử dụng ở mức liều tương ứng, mà chúng chỉ khác nhau về tần suất các tác dụng không mong muốn gặp phải, thế hệ 2 ít gặp hơn thế hệ 1. Do đó trên thực tế thế hệ 1 hiện nay hầu như không được sử dụng.
Các sulfonylureas được chuyển hóa tại gan, chất chuyển hóa có thể có hoặc không có hoạt tính. Thuốc được thải trừ qua thận (giảm liều ở bệnh nhân suy thận).
Nguy cơ gây hạ glucose huyết quá mức phụ thuộc vào thời gian bán thải (t1/2): Các thuốc có t1/2 dài như Chlorpropamide, Glyburide hay gây hạ đường huyết quá mức hơn, đặc biệt là ở bệnh nhân có nguy cơ hạ đường huyết cao. Những bệnh nhân này nên bắt đầu điều trị bằng các thuốc có t1/2 ngắn hơn và với liều ban đầu thấp hơn.
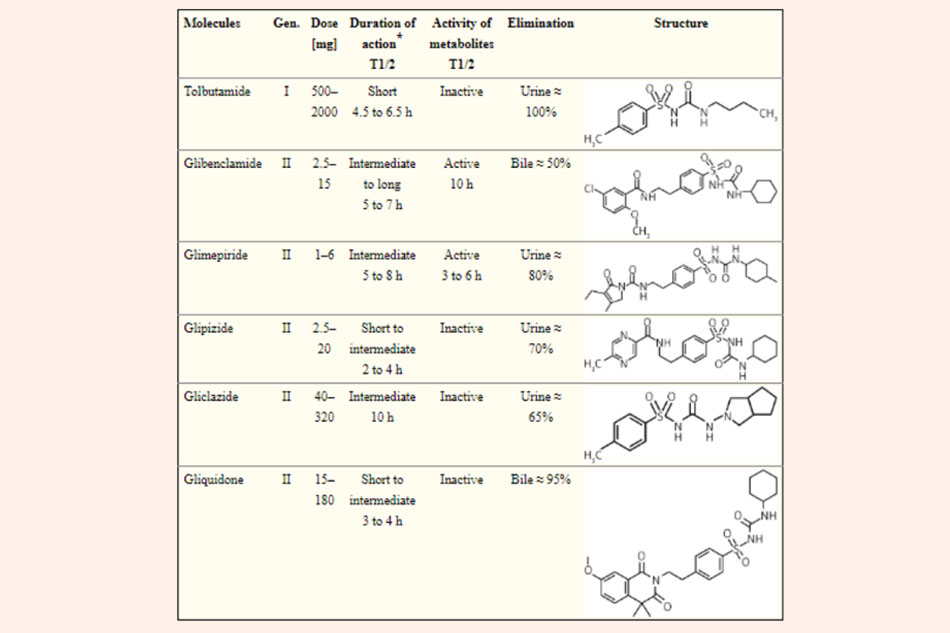
Thời gian tác dụng ngắn: < 12 giờ, trung bình: 12-24 giờ, dài: > 24 giờ.
Cơ chế tác dụng: Các sulfonylureas kích thích tế bào β đảo tụy tiết ra insulin nhiều hơn, do đó các thuốc này chỉ có tác dụng khi các tế bào β đảo tụy còn hoạt động. Ngoài ra, khi sử dụng thuốc, nó còn có hiệu ứng giảm sự chuyển hóa và thanh thải insulin ở gan. Do đó thuốc làm kéo dài tác dụng của insulin. Đặc biệt, tác dụng thứ hai này xuất hiện chủ yếu sau khi thuốc đã làm tăng bài tiết insulin. Trên thực tế, trong tháng đầu tiên điều trị, nồng độ insulin và đáp ứng với insulin tăng nhanh, dẫn đến hạ đường huyết. Sau giai đoạn này, nồng độ insulin cả khi cơ bản và khi kích thích trở nên thấp hơn so với nồng độ insulin định lượng được lúc bắt đầu điều trị, tuy nhiên, giá trị đường huyết không có sự thay đổi đáng kể. Lý do là gì chưa rõ.
Về cơ chế tác dụng ở cấp độ phân tử, các sulfonylureas liên kết với thụ thể đặc hiệu của chúng (SUR1) trên các tế bào β đảo tụy, ngăn chặn dòng ion kali (K+) đi qua kênh phụ thuộc ATP: Dòng ion K+ trong tế bào β không đi ra ngoài tế bào được, màng tế bào bị khử cực, do đó nó hoạt hóa mở kênh Ca2+ như mô tả ở hình dưới. Một lượng lớn ion Ca2+ đi vào tế bào làm các sợi actomyosin co lại, các sợi này chịu trách nhiệm xuất bào insulin (giải phóng insulin ra khỏi tế bào), do đó insulin được bài tiết ra với số lượng lớn.
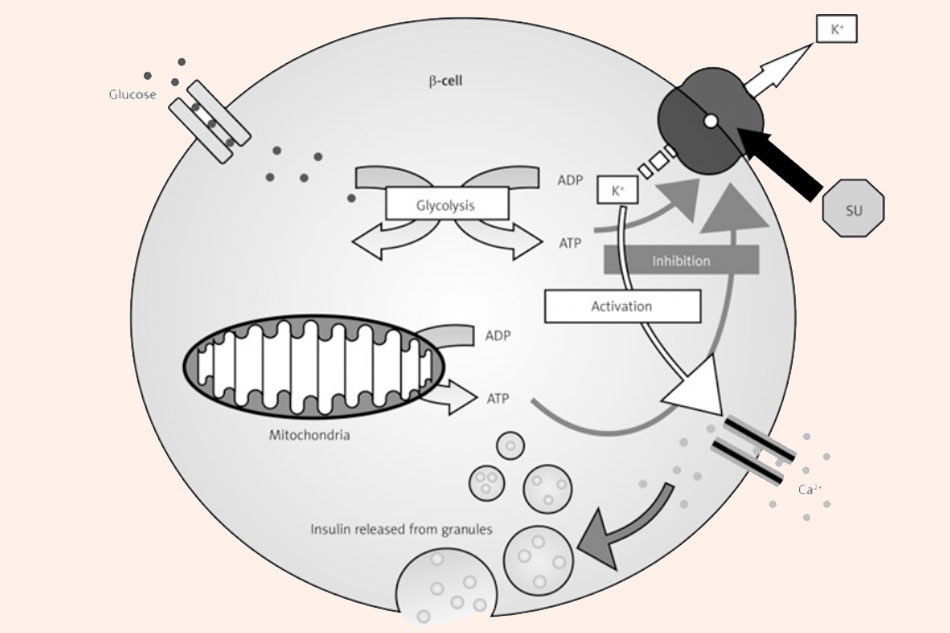
Các sulfonylureas có hiệu lực cao, giảm được HbA1c trung bình 1.5-2.0%, giảm được nồng độ glucose huyết tương lúc đói khoảng 60-70 mg/dL.
Các sulfonylureas có làm giảm biến chứng mạch máu nhỏ nhưng làm giảm biến chứng mạch máu lớn.
Chỉ định: Đái tháo đường type 2 (các tế bào β đảo tụy còn tiết được insulin). Thuốc thường được ưu tiên sử dụng cho những bệnh nhân có thể trạng gầy hoặc bình thường.
Phần lớn bệnh nhân chỉ sử dụng sulfonylureas đơn độc sẽ không thể đạt được sự kiểm soát đường huyết đầy đủ. Nguyên nhân thất bại có thể được chia ra 2 trường hợp:
- Không đáp ứng với sulfonylureas (giảm glucose huyết tương lúc đói < 30 mg/dL), nồng độ C-peptide thấp và nồng độ glucose huyết tương lúc đói cao (> 250 mg/dL), có dấu hiệu “nhiễm độc glucose” hoặc bệnh nhân chuyển sang đái tháo đường type 1.
- Đáp ứng tốt với sulfonylureas (giảm glucose huyết tương lúc đói > 30 mg/dL) nhưng không đạt được nồng độ glucose huyết tương mục tiêu. 75% bệnh nhân thất bại điều trị rơi vào nhóm này.
Các trường hợp thành công thường có nồng độ C-peptide cao, bệnh nhân mới mắc đái tháo đường type 2, không có đái tháo đường type 1, tăng glucose huyết tương lúc đói ở mức độ trung bình (< 250 mg/dL).
Kể cả khi có đạt được nồng độ glucose huyết tương mục tiêu thì thường sẽ có 5-7% bệnh nhân thất bại điều trị sau mỗi năm do các tế bào β đảo tụy bị kích thích tiết insulin quá nhiều nên suy kiệt dần.
Tác dụng không mong muốn:
- Hạ đường huyết quá mức: Những bệnh nhân có nồng độ glucose huyết tương lúc đói thấp hay bị tụt đường huyết hơn. Ngoài ra, tác dụng không mong muốn này còn hay gặp ở bệnh nhân bỏ bữa, ăn uống thất thường, vận động nặng, giảm cân nhiều.
- Tăng cân: Do kích thích tăng tiết insulin làm tăng quá trình đồng hóa. Tác dụng không mong muốn này không thể tránh khỏi.
- Hạ natri máu (nồng độ Na+ huyết thanh < 129 mEq/L): Thường gặp nhất khi bệnh nhân dùng Chlorpropamide (5%) và Tolbutamide, đặc biệt khi phối hợp chúng với các thuốc lợi tiểu (gây mất natri qua nước tiểu).
Chống chỉ định: Quá mẫn cảm với sulfonylureas hoặc bất cứ thành phần nào của thuốc. Phụ nữ mang thai. Người bị đái tháo đường type 1.
Thận trọng với bệnh nhân suy giảm chức năng gan thận.
Meglitinides:
Các thuốc nhóm này được gọi là các “glinides”, bao gồm: Repaglinide, Nateglinide… Các chất này còn được gọi là “Meglitinide analogues”, được giới thiệu vào năm 1995 và được chấp thuận sử dụng lâm sàng ở người trưởng thành mắc đái tháo đường type 2 vào năm 2000.
Các thuốc này có đặc điểm chung là hấp thu nhanh, thời gian bán thải (t1/2) ngắn (Repaglinide 0.5-1 giờ, Nateglinide 1-1.5 giờ). Các thuốc này chỉ được sử dụng để tăng tiết insulin sau ăn. Tác dụng tương tự như sulfonylureas nhưng yếu hơn.
Cơ chế tác dụng: Thuốc kích thích tăng cường bài tiết insulin, cơ chế của chúng là gắn vào các kênh kali (K+) phụ thuộc ATP và gây ra tác dụng tương tự như các sulfonylureas. Mặc dù tạo ra tác dụng tương tự và có chế khá tương đồng với các sulfonylureas, nhưng các thuốc nhóm này vẫn được phân ra thành một nhóm riêng vì chúng gắn vào vị trí khác trên kênh K+ so với các sulfonylureas.
Chỉ định: Đái tháo đường type 2, kiểm soát tăng đường huyết sau ăn.
Hiện nay các thuốc trong nhóm này không được sử dụng phổ biến.
Tác dụng không mong muốn: Hạ glucose huyết quá mức: Tác dụng không mong muốn này có thể xảy ra nhưng ít hơn các sulfonylureas.
Chống chỉ định: Quá mẫn cảm với hoạt chất hoặc bất kỳ thành phần nào của thuốc. Bệnh nhân đái tháo đường type 1. Repaglinide chống chỉ định ở những bệnh nhân cũng đang dùng đồng thời Gemfibrozil.
Biguanides:
Metformin là một trong những loại thuốc được sử dụng rộng rãi nhất trên thế giới để điều trị đái tháo đường type 2 vì những ưu điểm vượt trội của nó: giá thành rẻ, hiệu lực cao, có thể sử dụng lâu dài, có hiệu quả kể cả khi sulfonylureas đã không còn tác dụng. Đây là thuốc duy nhất trong nhóm biguanides còn được sử dụng cho đến ngày nay. Còn lại thuốc khác trong nhóm đã bị ngừng vĩnh viễn do nguy cơ gây nhiễm toan lactic nặng, đe dọa tính mạng.
Các biguanides đầu tiên là Metformin và Phenformin (Phenformin sau này đã bị thu hồi tại nhiều quốc gia vì tác dụng phụ gây nhiễm toan lactic nghiêm trọng, hiện nay chúng ta không thể tìm thấy thuốc này trên thị trường được nữa, nhưng đã có những trường hợp tại Việt Nam trộn Phenformin vào các sản phẩm có nguồn gốc từ dược liệu và bán cho những bệnh nhân đái tháo đường) có cấu trúc khung được xuất phát từ galegine, một hoạt chất tự nhiên có nguồn gốc từ cây Galega officinalis, loài cây này được sử dụng làm thuốc thảo dược ở châu Âu thời trung cổ. Năm 1920, galegine được thử nghiệm tác dụng hạ glucose máu trên người, tuy nhiên sau đó thử nghiệm đã thất bại vì người ta nhận ra galegine quá độc để có thể sử dụng. Gần như vào cùng khoảng thời gian đó, hai dẫn chất của galegine là Metformin và Phenformin, lần đầu tiên đã được tổng hợp hóa học và đã được thử nghiệm, cho thấy tác dụng hạ đường huyết rất tốt. Nhưng phải mãi cho đến những năm 1950, chúng mới được đưa vào sử dụng trên lâm sàng.
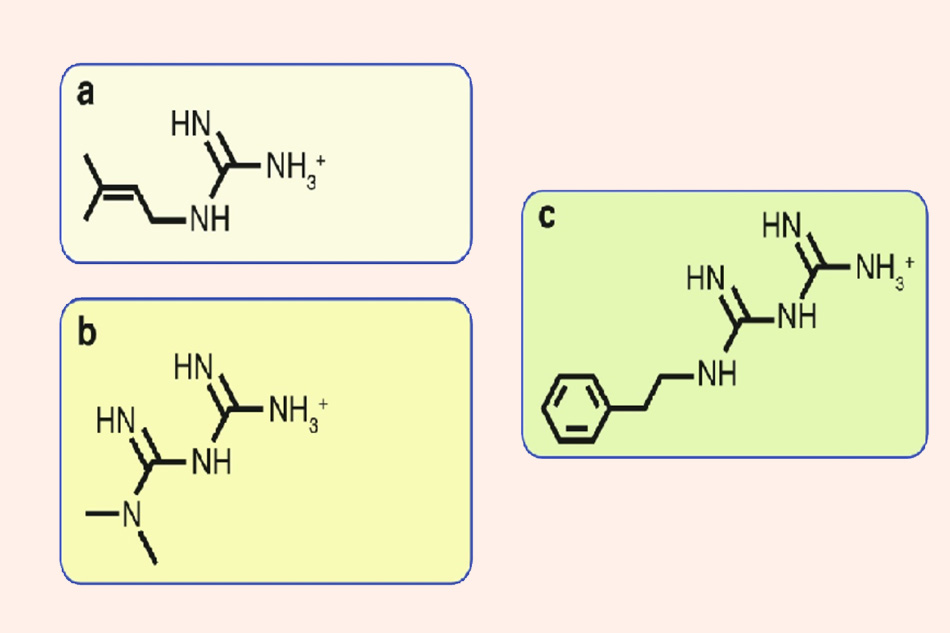
Đây là công thức cấu tạo của galegine (a), Metformin (b) và Phenformin (c). Metformin và Phenformin là các dẫn chất tổng hợp hóa học của galegine. Galegine còn có tên gọi khác là isoprenylguanidine, là một dẫn chất isoprenyl của guanidine, trong khi đó, Metformin (hay còn gọi là dimethylbiguanide) và Phenformin (hay còn gọi là phenethylbiguanide) là hai dẫn chất thế của nó.
Metformin có sinh khả dụng (F) khoảng 50-60%. Metformin không bị chuyển hóa và không liên kết với protein huyết tương. Thải trừ qua thận theo cơ chế lọc ở cầu thận và bài tiết ở ống thận, thời gian bán thải (t1/2) khoảng 6 giờ. Tác dụng kéo dài trên 24 giờ.
Metformin làm giảm HbA1c khoảng 1.5-2.0% (hiệu lực cao), giảm nồng độ glucose huyết tương lúc đói khoảng 60-80 mg/dL. Không giống như các sulfonylureas, Metformin có thể giảm glucose huyết tương lúc đói ngay cả khi nó rất cao (> 300 mg/dL) hoặc khi có tình trạng kháng insulin nặng.
Điều đặc biệt ở Metformin là thuốc có tác dụng làm giảm triglyceride và LDL-C từ 8-15%, tăng HDL-C nhẹ (2%). Đây đều là các tác dụng có lợi cho tim mạch.
Metformin cũng làm giảm cân nhẹ (2-3 kg), điều này đặc biệt có ý nghĩa vì phần đa các bệnh nhân mắc đái tháo đường type 2 có thừa cân hoặc béo phì.
Tác dụng làm giảm biến chứng mạch máu nhỏ của metformin tương đương với Insulin và các sulfonylureas.
Metformin không làm giảm biến chứng mạch máu lớn trên các bệnh nhân có cân nặng bình thường, nhưng làm giảm biến chứng mạch máu lớn trên các bệnh nhân có thừa cân hoặc béo phì. Metformin làm giảm rõ rệt tỷ lệ đột quỵ và tử vong do biến cố tim mạch, giảm tỷ lệ tử vong do đái tháo đường và giảm tỷ lệ nhồi máu cơ tim so với các sulfonylureas và Insulin.
Cơ chế tác dụng: Metformin là một loại thuốc có cơ chế tác dụng rất phức tạp và đa dạng, với nhiều tác động lên nhiều cơ quan khác nhau. Trên gan, Metformin làm giảm sản xuất glucose, điều này làm hạ glucose máu. Tác động này có thể là trực tiếp hoặc gián tiếp. Trên ruột, Metformin kích thích các tế bào đặc hiệu ở đây tiết ra GLP-1 (glucagon-like peptide-1), điều này cũng làm tăng tiết insulin làm hạ glucose máu, đồng thời tạo ra sự thay đổi trong hệ vi sinh đường ruột.
Về cơ chế ở mức độ phân tử, Metformin ức chế chuỗi hô hấp của ty thể trong tế bào gan, điều này làm hoạt hóa AMPK, tăng cường sự nhạy cảm insulin (thông qua các tác động có lợi trên chuyển hóa lipid) và làm giảm AMP vòng (cAMP), từ đó làm giảm biểu hiện của enzyme tân tạo đường (giảm tân tạo đường thì nồng độ glucose máu giảm). Metformin cũng có tác dụng độc lập với AMPK trên gan nhờ hoạt hóa theo cơ chế lysosome, có thể ức chế fructose-1,6-bisphosphatase nhờ làm tăng tỷ lệ AMP/ATP, từ đó cũng làm giảm nồng độ glucose máu do giảm tân tạo đường (xem hình dưới).
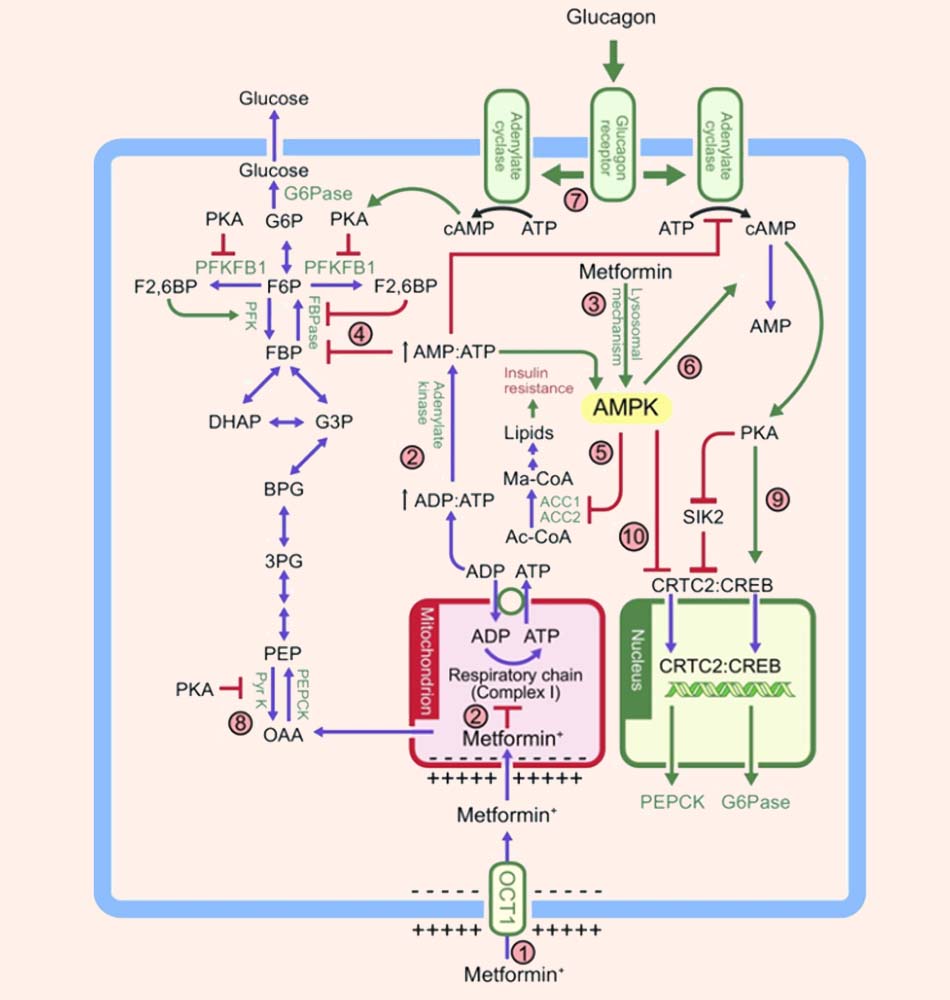
Metformin ảnh hưởng đến chuyển hóa trong tế bào gan thông qua nhiều cơ chế tác dụng. (1) Metformin được vận chuyển vào tế bào gan nhờ sự xúc tác của chất vận chuyển cation hữu cơ-1 (OCT1). Metformin là phân tử tích điện dương (là cơ chất của chất vận chuyển OCT1) và đồng thời, Metformin cũng có thể di chuyển vào trong ty thể vì cấu trúc màng sinh chất và màng trong của ty thể có sự tương đồng. (2) Metformin ức chế Phức hệ I trong chuỗi hô hấp tế bào, ức chế sự sản xuất ATP trong ty thể và do đó, thuốc làm tăng tỷ lệ ADP/ATP và AMP/ATP trong bào tương (sau này còn có cơ chế hoạt hoá adenylate kinase), tất cả những tác động nói trên hoạt hóa AMPK. (3) AMPK có thể được hoạt hóa theo cách khác bằng cơ chế lysosome, nhưng cơ chế cụ thể không được trình bày chi tiết trong hình trên. (4) Tỷ lệ AMP/ATP tăng cũng có tác dụng ức chế fructose-1,6-bisphosphatase (FBPase), dẫn đến ức chế tân tạo đường, đồng thời adenylate cyclase cũng bị ức chế và làm giảm sản xuất cAMP. (5) AMPK đã hoạt hóa theo cơ chế phosphoryl hóa các đồng phân ACC1 và ACC2 của ACC, ức chế quá trình tổng hợp lipid và thúc đẩy quá trình oxy hóa lipid, do đó làm giảm dự trữ lipid trong gan và giảm sự đề kháng với insulin của gan. (6) AMPK cũng phosphoryl hóa và hoạt hóa phosphodiesterase 4B (PDE4B) vòng 3′,5′ đặc hiệu cAMP, do đó làm tăng chuyển cAMP thành AMP, gây giảm cAMP. (7) Mô tả glucagon liên kết với receptor trên màng tế bào gan của nó, thông qua adenylate cyclase, chuyển ATP thành cAMP, điều này hoạt hóa protein kinase A (PKA) phụ thuộc cAMP, PKA lại gây ra phosphoryl hóa và bất hoạt PFKFB1, làm giảm nồng độ fructose-2,6-bisphosphate (F2,6BP), F2,6BP lại là một chất hoạt hóa phosphofructokinase (PFK) theo cơ chế dị lập thể và ức chế fructose-1,6-bisphosphatase (FBPase). Kết quả cuối cùng glucagon dẫn đến tăng tân tạo đường, làm tăng glucose máu. (8) PKA cũng phosphoryl hóa và làm bất hoạt enzyme pyruvate kinase (Pyr K), chịu trách nhiệm cho quá trình đường phân trong tế bào gan. (9) PKA cũng phosphoryl hóa yếu tố phiên mã gắn yếu tố đáp ứng cAMP (CREB), gây ra sự cảm ứng phiên mã của các gen mã hóa các enzyme tân tạo đường PEPCK và G6Pase, làm tăng đường huyết. (10) AMPK phosphoryl hóa chất đồng hoạt hóa phiên mã điều hòa bởi CREB-2 (CRTC2), hoặc phosphoryl bởi các kinases liên quan đến AMPK như kinase cảm ứng muối 2 (SIK2), khiến CRTC2 bị giữ lại trong tế bào chất, đối kháng với tác dụng của PKA trên phiên mã PEPCK và G6Pase, làm giảm sản xuất PEPCK và G6Pase. PKA ức chế SIK2 bằng cách phosphoryl hóa trực tiếp tại nhiều vị trí của nó.
Các từ ngữ viết tắt: Ac-CoA: acetyl-CoA, Ma-CoA: malonyl-CoA, BPG: 1,3-bisphosphoglycerate, 3PG: 3-phosphoglycerate, FBP: fructose 1,6-bisphosphate, DHAP: dihydroxyacetone phosphate, G3P: glyceraldehyde 3-phosphate, G6P: glucose 6-phosphate, F6P: fructose 6-phosphate, OAA: oxaloacetate, PEP: phosphoenolpyruvate.
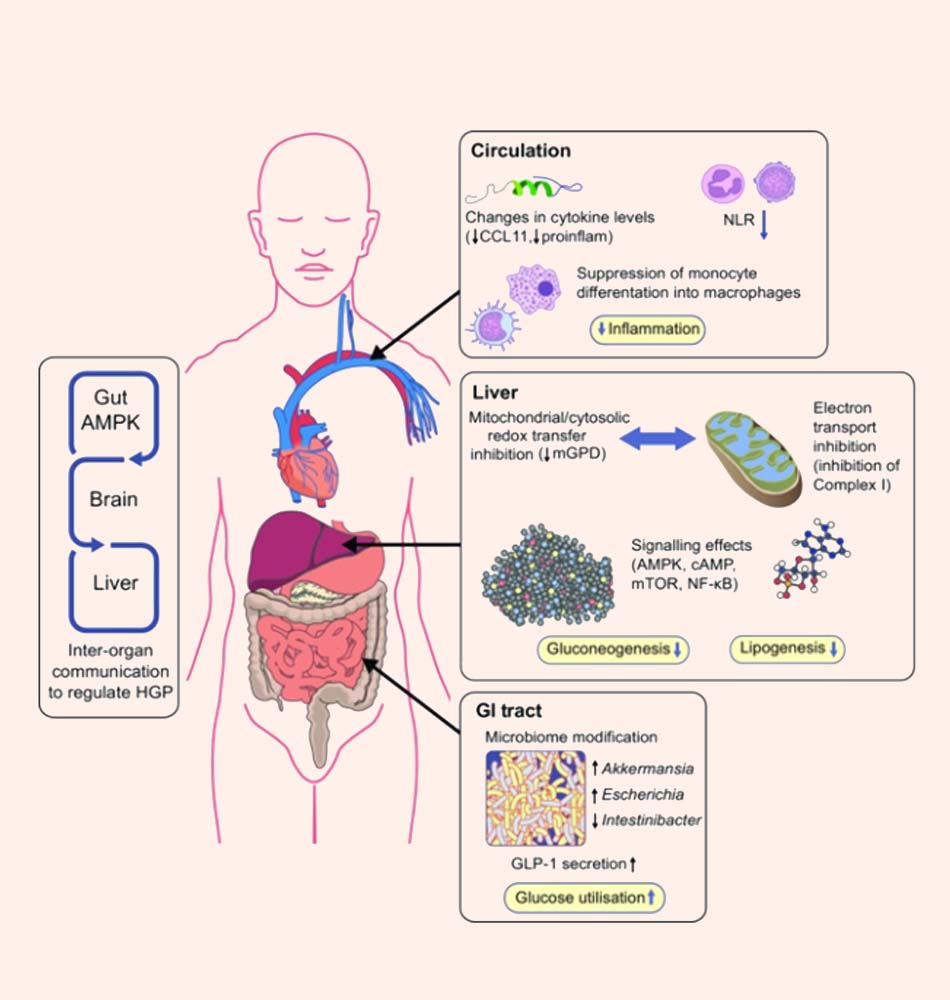
Tác động của Metformin trên quá trình chuyển hóa và đáp ứng viêm. Trong các nghiên cứu quan sát (observational studies), bệnh nhân mắc đái tháo đường type 2 thường có chỉ số NLR (tỷ lệ bạch cầu trung tính/bạch cầu lympho) thấp hơn người bình thường. Trong các thử nghiệm đối chứng ngẫu nhiên (randomized controlled trials), nhiều cytokines đã được chứng minh là bị ức chế bởi Metformin, trong đó có bao gồm cả C-C motif chemokine 11 (CCL11, còn được biết đến với tên gọi là eotaxin-1). Nhiều kết quả từ các nghiên cứu khác nhau cho thấy Metformin ức chế sự biệt hóa các tế bào bạch cầu đơn nhân thành đại thực bào, ức chế sự bài tiết các cytokines tiền viêm. Trên một số tế bào ruột, Metformin cho thấy tác dụng kích thích bài tiết GLP-1 (một loại incretin làm tăng tiết insulin) và làm thay đổi hệ vi sinh đường ruột. Cũng có những bằng chứng cho thấy ảnh hưởng của Metformin qua trung gian ruột thông qua con đường ruột – não – gan, từ đó điều hòa lượng glucose được sản xuất trong gan một cách gián tiếp, nhưng số lượng bằng chứng chưa nhiều. Như đã nói ở hình trên, Metformin làm giảm quá trình tổng hợp lipid và tân tạo đường ở các tế bào gan, thông qua một loạt các cơ chế phức tạp ở cấp độ phân tử.
Các từ ngữ viết tắt: HGP: sản xuất glucose ở gan, mTOR: đích của Rapamycin ở động vật có vú.
Chỉ định: Đái tháo đường type 2.
Tác dụng không mong muốn:
- Rối loạn tiêu hóa: Buồn nôn, tiêu chảy xảy ra ở khoảng 30% bệnh nhân. Để khắc phục tác dụng không mong muốn này, cần dùng bắt đầu từ liều nhỏ để bệnh nhân quen dần, sau đó tăng dần liều. Liều tối đa thường là 2 g/ngày.
- Khác: Chán ăn, đầy bụng (điều này có thể liên quan đến tác dụng giảm cân của Metformin).
- Lưỡi có vị kim loại, giảm hấp thu vitamin B12, tụt glucose huyết tương khi vận động quá nhiều: Ít gặp trên lâm sàng.
- Nhiễm toan lactic: Nguyên nhân là do glucose tăng chuyển hóa theo con đường tạo ra acid lactic. Tỷ suất gặp phải tác dụng không mong muốn này là trung bình 3 case/100,000 người mỗi năm.
Chống chỉ định: Quá mẫn cảm với bất cứ thành phần nào của thuốc. Phụ nữ có thai, người suy gan, thận nặng, suy tim, suy hô hấp.
Thuốc ức chế α-glucosidase:
Các thuốc trong nhóm này có thể kể đến bao gồm Acarbose, Voglibose và Miglitol. Các thuốc này hiện nay cũng không dùng nhiều trong điều trị đái tháo đường.
Acarbose là loại thuốc được sử dụng nhiều nhất trong nhóm này tại Việt Nam. Nó được chiết xuất từ quá trình lên men của một loài vi sinh vật có tên là Actinoplanes utahensis.
Về dược động học, Acarbose gần như không được hấp thu ở ruột non vì nó hầu như không thể bị phá hủy ở ruột non bởi các enzyme tiêu hóa, nó được bài xuất qua phân và phần lớn ở dưới dạng nguyên vẹn, không được chuyển hóa. Tuy nhiên có đến khoảng 30% thuốc bị chuyển hóa. Lý do không phải là do cơ thể chúng ta, mà là do các vi sinh vật sinh sống tại đại tràng đã lên men chúng. Cũng chính vì điều này mà Acarbose cũng gây ra tác dụng phụ thường gặp của nó là tiêu chảy.
Voglibose có các thông số dược động học gần tương tự như Acarbose. Nó cũng được hấp thu rất kém tại ruột non và bị đào thải nhanh chóng ra ngoài theo đường phân. Hiện nay, các nhà khoa học vẫn chưa xác định được các chất chuyển hóa của Voglibose bởi vi sinh vật trong đại tràng.
Hoàn toàn ngược lại với 2 loại thuốc được nêu trên, dược động học của Miglitol có hấp thu hoàn toàn trong ruột non và thải trừ dưới dạng không đổi qua thận!
Các thuốc này làm giảm glucose huyết tương sau ăn (40-50 mg/dL), giảm HbA1c 0.3-1.0%. Do đó những bệnh nhân có HbA1c và nồng độ glucose huyết tương lúc đói gần với ngưỡng mục tiêu nhưng glucose huyết tương sau ăn cao thì phù hợp với thuốc này.
Thuốc làm giảm nhẹ biến chứng mạch máu nhỏ và cả biến chứng mạch máu lớn.
Cơ chế tác dụng: Các thuốc nhóm này là các carbohydrates giả có khả năng ức chế cạnh tranh các enzyme α-glucosidase nằm trên diềm bàn chải các tế bào ruột, các enzyme này chịu trách nhiệm thủy phân các oligosaccharides và polysaccharides (các carbohydrates phức tạp) vốn dĩ không thể hấp thu thành các monosaccharides đơn giản mà cơ thể có thể hấp thu được. Do vậy thuốc không có tác dụng hạ đường huyết trực tiếp mà chỉ có tác dụng chống tăng đường huyết sau ăn.
Acarbose là một tetrasaccharide (chứa 4 đơn phân [monomer] đường) giả với một nguyên tử nitơ liên kết giữa đơn vị glucose thứ nhất và thứ hai trong cấu trúc hóa học. Acarbose có ái lực cao đối với các trung tâm hoạt động của α-glucosidase ở diềm bàn chải của ruột non, đồng thời nó cũng rất ổn định về mặt hóa học. Hiệu lực ức chế cao nhất của Acarbose là đối với với enzyme glucoamylase, tiếp theo là các enzyme sucrase, maltase và dextranase. Nó cũng có thể ức chế được α-amylase tuy rằng hiệu lực trên enzyme này yếu hơn, và nó không có tác dụng ức chế các β-glucosidase, ví dụ như enzyme lactase.
Chỉ định: Đái tháo đường type 2, chống tăng glucose huyết tương sau ăn.
Vì các thuốc ức chế α-glucosidase ức chế sự thủy phân và tiêu hóa các carbohydrate phức tạp, các loại thuốc này nên được uống vào thời điểm bắt đầu bữa ăn chính, uống cùng với miếng đầu tiên của bữa ăn. Nếu ăn xong mới uống thuốc thì khi đó các carbohydrate phức tạp đã bị thủy phân gần hết và việc sử dụng các thuốc này sẽ không còn ý nghĩa nữa. Thêm vào đó, lượng carbohydrate phức tạp trong bữa ăn sẽ quyết định hiệu quả của các loại thuốc này trong việc giảm glucose huyết tương sau ăn, vì chúng hoạt động theo cơ chế ức chế cạnh tranh enzyme tại ruột, nên lượng carbohydrate phức tạp mà bệnh nhân ăn vào trong bữa ăn càng nhiều thì hiệu quả ức chế enzyme của thuốc càng giảm.
Tác dụng không mong muốn: Tác dụng không mong muốn phổ biến hay gặp nhất với Acarbose và Voglibose là tiêu chảy do sự lên men thuốc tại đại tràng bởi các vi sinh vật cộng sinh. Để tránh tác dụng không mong muốn này, nên bắt đầu sử dụng thuốc với liều nhỏ, sau đó tăng dần liều lên đến liều điều trị cần thiết. Ngoài ra, thuốc có thể làm giảm hấp thu sắt.
Chống chỉ định:
Chống chỉ định Acarbose và Voglibose cho các trường hợp:
- Phụ nữ có thai và phụ nữ đang cho con bú.
- Viêm ruột, đặc biệt ở những bệnh nhân đang có loét.
- Bệnh nhân bị suy gan hoặc có tăng men gan.
- Người bị hạ đường huyết.
- Bệnh nhân có nhiễm toan chuyển hóa (ví dụ như biến chứng của đái tháo đường type 1).
Thiazolidinediones:
Các thiazolidinediones thường có tên gọi là các “glitazone”, trước đây chúng đã từng là các thuốc bom tấn và mang lại nhiều triển vọng về một loại thuốc mới điều trị đái tháo đường type 2 thay thế cho các sulfonylureas hoặc Insulin. Nhưng sau khi tung ra thị trường, nhiều nhược điểm của các thuốc nhóm này mới bắt đầu bộc lộ trong các nghiên cứu hậu mãi. Chính vì vậy mà hiện nay nhóm này chỉ còn duy nhất một thuốc còn được sử dụng rộng rãi trên thị trường Hoa Kỳ là Pioglitazone, còn Rosiglitazone đã bị hạn chế sử dụng hơn rất nhiều, Troglitazone thì đã bị thu hồi từ những năm 2000 do nguy cơ gây nhiễm độc gan. Hiện tại các thuốc này cũng không phổ biến ở Việt Nam.
Các thuốc nhóm này có tác dụng hạ đường huyết chậm, thường mất từ 2-3 tháng để có tác dụng tối đa, do vậy phải sử dụng thuốc kéo dài. Do đó nếu như sau 3 tháng sử dụng thuốc này mà vẫn không đạt được đáp ứng đáng kể trên trên sàng, nên xem xét đổi thuốc.
Hiệu lực: Các thuốc nhóm này đều hạ HbA1c ở mức độ trung bình (0.5-1.5%).
Điều trị kéo dài với các thuốc nhóm này làm tăng HDL-C, LDL-C và giảm triglyceride huyết thanh.
Tất cả các thuốc nhóm này đều được chuyển hóa qua gan qua hệ thống CYP450, cần chú ý tương tác thuốc có thể xảy ra.
Cơ chế tác dụng: Các thiazolidinediones là thuốc chủ vận receptor nhân PPAR-γ. Cơ chế thực sự còn nhiều điểm chưa rõ ràng. Hoạt hóa PPAR-γ làm tăng sản xuất adiponectin, làm tăng hoạt động của insulin. Hoạt hóa PPAR-γ cũng tác động lên hệ vận chuyển glucose GLUT4, tăng cường hấp thu và sử dụng glucose trong tế bào, đồng thời làm các tế bào mô mỡ nhạy cảm hơn với insulin.
Thuốc gây phân bố lại mỡ, giảm mỡ nội tạng và tăng mỡ dưới da. Dùng thuốc đơn độc không gây tăng cân, không gây hạ đường huyết quá mức.
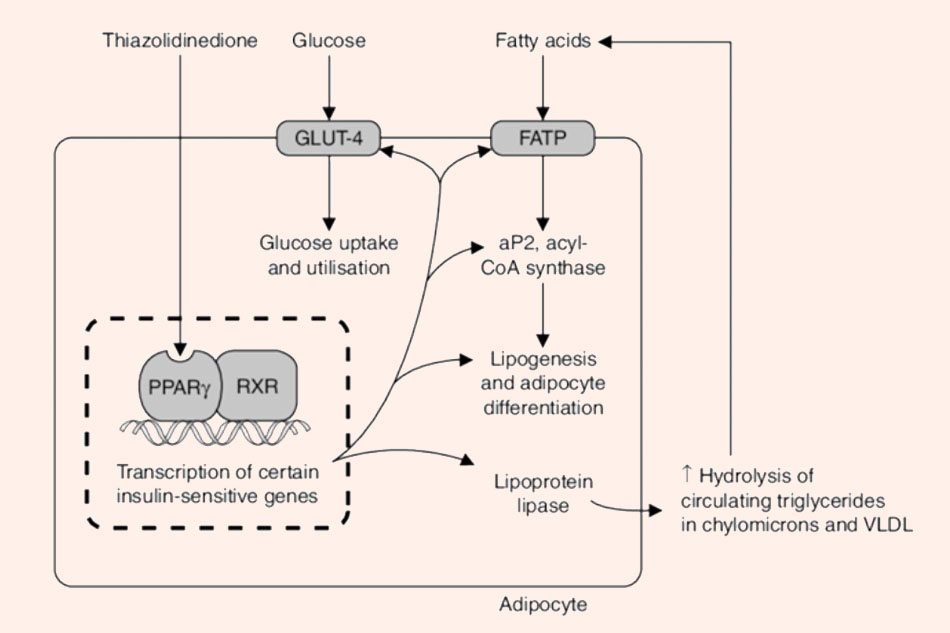
Chỉ định: Đái tháo đường type 2.
Tác dụng không mong muốn:
- Thiếu máu nhẹ.
- Tổn thương gan: Có thể dẫn đến suy gan không hồi phục, tử vong, đặc biệt là Troglitazone. Pioglitazone và Rosiglitazone ít độc hơn.
- Ung thư bàng quang: Tất cả các thuốc trong nhóm này đều làm tăng nguy cơ gây ung thư bàng quang khi sử dụng trên 1 năm. FDA (Cơ quan Quản lý Thực phẩm và Dược phẩm) Hoa Kỳ không khuyến cáo sử dụng thuốc trên bệnh nhân ung thư bàng quang hoặc có tiền sử ung thư bàng quang.
- FDA đã cảnh báo về việc sử dụng Rosiglitazone làm tăng nguy cơ thiếu máu cục bộ cơ tim so với giả dược, Metformin hay sulfonylureas. Đáng chú ý là thuốc cùng nhóm Pioglitazone lại làm giảm nguy cơ tim mạch.
Chống chỉ định: Quá mẫn cảm với bất cứ thành phần nào của thuốc. Phụ nữ có thai, bệnh nhân ung thư bàng quang hoặc có tiền sử ung thư bàng quang, người suy gan nặng.
Thuốc chủ vận thụ thể GLP-1:
Thuốc chủ vận thụ thể GLP-1 là một thuốc ít phổ biến ở Việt Nam nhưng đã phổ biến ở nhiều nước trên thế giới, thực sự hiệu quả trong điều trị đái tháo đường type 2, và đồng thời đi kèm với đó là giá cả điều trị không hề rẻ.
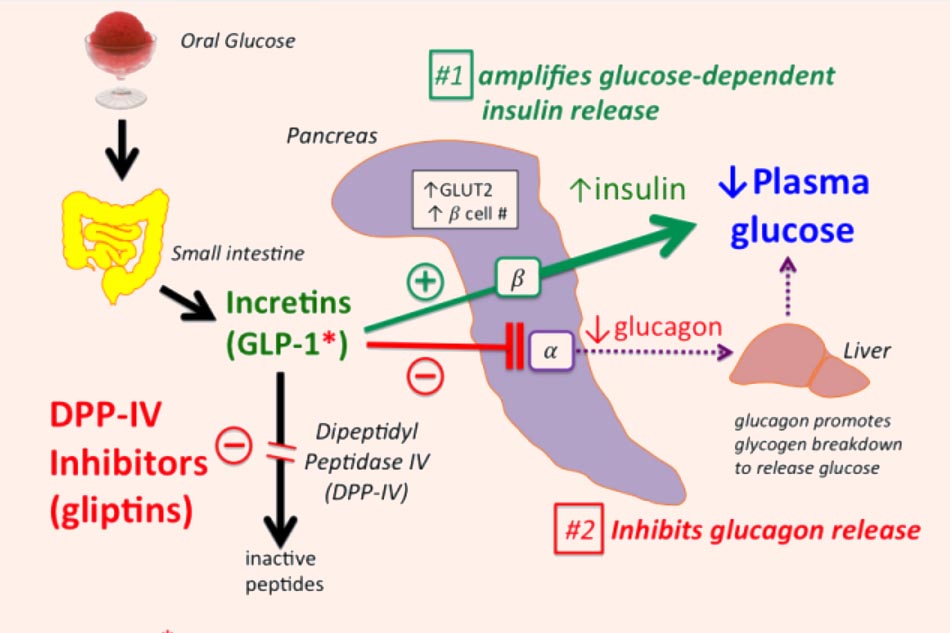
Đầu tiên chúng ta cần tìm hiểu về incretin. Đây là một hormon tại chỗ, lần đầu tiên được đưa ra định nghĩa bởi La Barre, là các chất có khả năng hạ glucose huyết được tìm thấy trong dịch chiết tá tràng. Năm 1962, Elrick và các cộng sự, McIntyre và các cộng sự, đã báo cáo độc lập với nhau về hiện tượng dùng glucose đường uống làm tăng tiết insulin đáng kể hơn so với dùng glucose đường tĩnh mạch, từ đó khái niệm “hiệu ứng incretin” ra đời. Yếu tố gây ra hiệu ứng incretin này có thể bắt nguồn từ ruột non.
Như vậy incretin là những hormone tại chỗ có bản chất peptide, được tiết vào máu vài phút sau khi thức ăn tác động lên niêm mạc ruột non và kích thích tế bào β đảo tụy bài tiết insulin. Hiện nay người ta đã nghiên cứu nhiều nhất về 2 incretin chính: GLP-1 (peptide giống glucagon 1, phát hiện năm 1983) và GIP (polypeptide hướng insulin phụ thuộc glucose, phát hiện năm 1970). Không chỉ kích thích bài tiết insulin, chúng còn làm chậm tốc độ tháo rỗng dạ dày và ức chế sự ngon miệng. GLP-1 được sản xuất bởi các tế bào L ở hồi tràng và đại tràng, còn GIP được sản xuất bởi các tế bào K ở tá tràng và hỗng tràng. Các hormone này đều có thời gian bán thải ngắn (vài phút) và bị phá hủy bởi enzyme Dipeptidyl Peptidase-4 (DPP-4).
Nhiều nghiên cứu cho thấy ở bệnh nhân đái tháo đường type 2, họ dường như không có hiệu ứng incretin hoặc có nhưng rất yếu. Vì GLP-1 có nhiều ưu điểm vượt trội hơn GIP, nên các nhà khoa học đã tập trung vào nghiên cứu các thuốc có cấu trúc tương tự GLP-1. Để khắc phục thời gian bán thải (t1/2) quá ngắn của GLP-1, các nhà khoa học đã thay đổi cấu trúc của GLP-1 người, hoặc dựa trên cấu trúc một chất chủ vận thụ thể GLP-1 có trong tự nhiên nhưng bền hơn với DPP-4, đó là exendin-4.
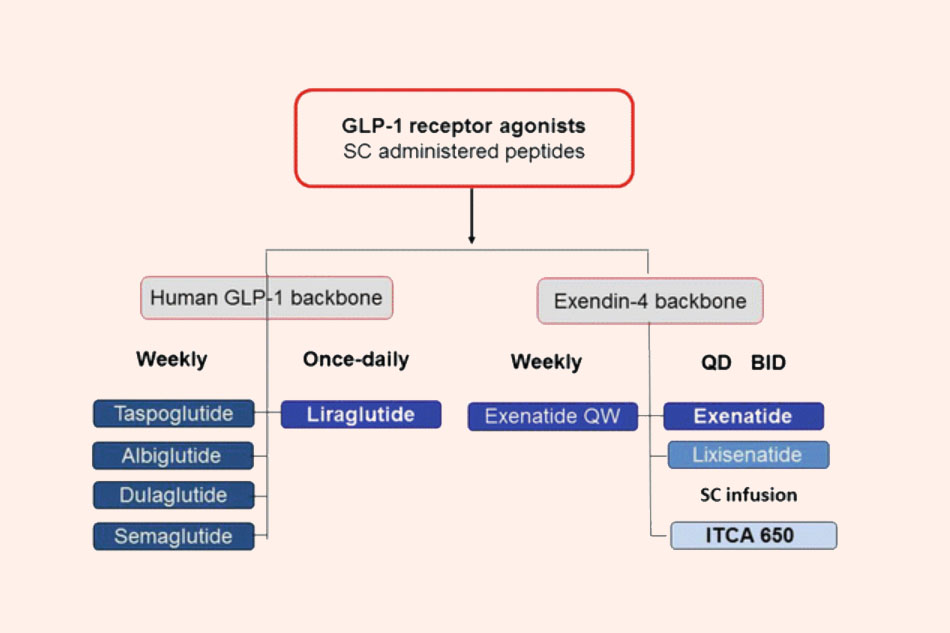
Đây là các thuốc chủ vận thụ thể GLP-1. Trái: Khung GLP-1 của người. Phải: Khung exendin-4.
Tùy theo thời gian tác dụng mà tần suất dùng thuốc khác nhau.
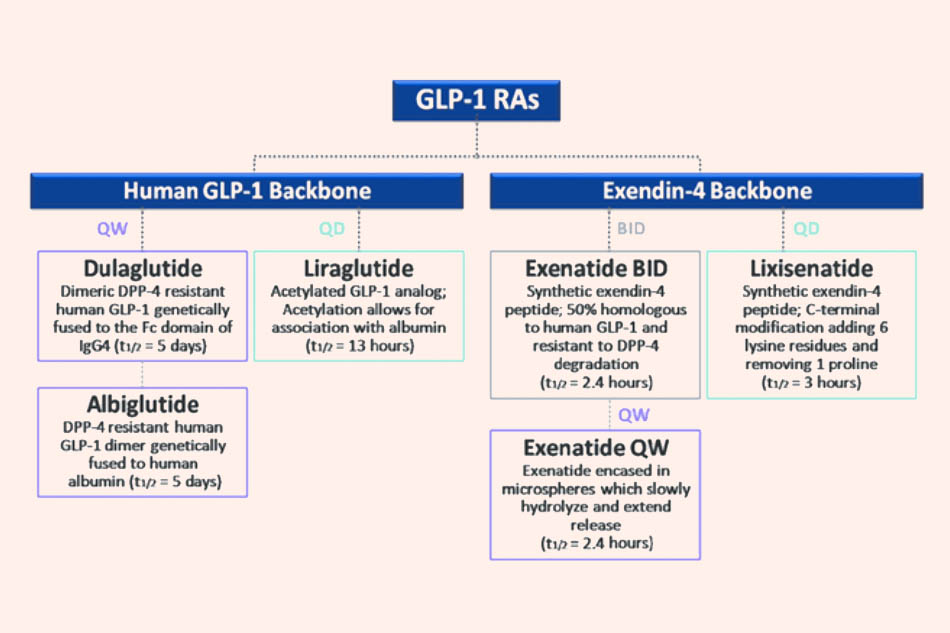
Đây là tần suất dùng thuốc của một số thuốc chủ vận thụ thể GLP-1. BID: 2 lần/ngày, QD: 1 lần/ngày, QW: 1 lần/tuần.
Hiệu lực: Các thuốc chủ vận thụ thể GLP-1 tác dụng kéo dài làm giảm HbA1c 0.9-1.9%.
Thuốc cũng làm giảm cân nhẹ (2-4 kg).
Đường dùng là tiêm dưới da (tương tự Insulin). Chi phí điều trị bằng các thuốc này không hề rẻ. Do đó đây không phải là các thuốc được dùng phố biến tại Việt Nam.
Cơ chế tác dụng: Thuốc bắt chước tác dụng của GLP-1 lên các tế bào có thụ thể GLP-1 và gây ra các tác dụng: tăng tiết insulin, ức chế tiết glucagon (đều phụ thuộc glucose), ức chế ngon miệng và làm chậm tốc độ tháo rỗng dạ dày.
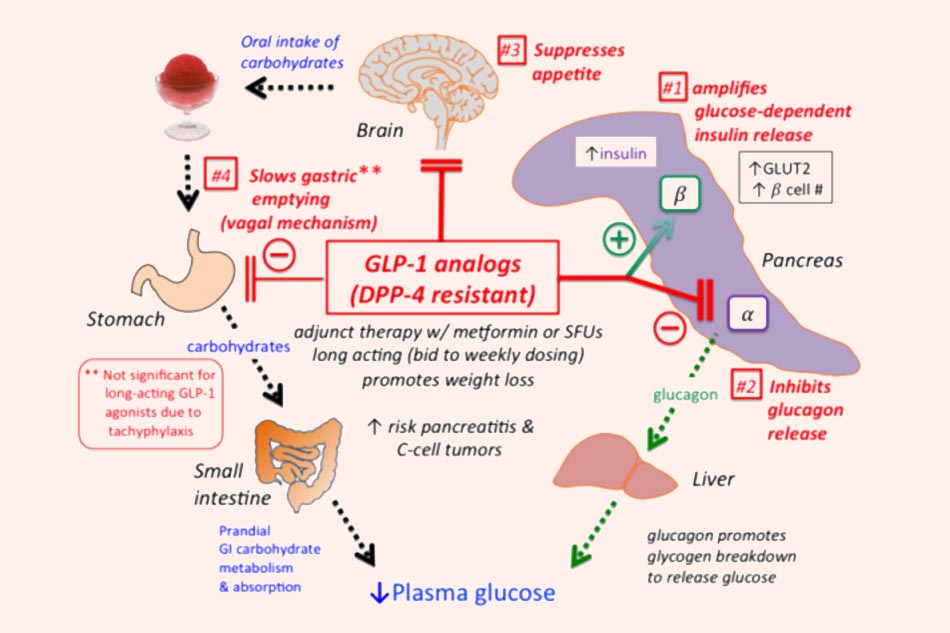
Chỉ định: Đái tháo đường type 2.
Tác dụng không mong muốn:
- Rối loạn tiêu hóa: Buồn nôn, nôn, tiêu chảy thường gặp.
- Cảnh báo: Viêm tụy, carcinoma tuyến giáp tủy (trừ Exenatide). Thận trọng khi sử dụng trên bệnh nhân có tiền sử hoặc nguy cơ viêm tụy.
- Do làm chậm tốc độ tháo rỗng dạ dày nên có thể ảnh hưởng đến hấp thu nhiều thuốc. Các thuốc dùng cùng có thể phải dùng trước 1 giờ hoặc sau 4 giờ khi dùng các thuốc chủ vận thụ thể GLP-1.
- Hạ đường huyết quá mức: Đặc biệt khi phối hợp với các Insulin, sulfonylureas.
- Phản ứng tại chỗ tiêm: Do thuốc làm tăng đáp ứng miễn dịch.
Chống chỉ định: Quá mẫn cảm với bất cứ thành phần nào của thuốc. Suy thận nặng (CrCl < 30 mL/phút). Viêm ruột, liệt dạ dày. Carcinoma tuyến giáp tủy và đa u nội tiết type 2 (trừ Exenatide). Phụ nữ mang thai. Phụ nữ đang cho con bú không được khuyến cáo dùng thuốc.
Thuốc ức chế DPP-4:
Sự ra đời của các thuốc ức chế DPP-4, còn được gọi là các “gliptin”, cũng có gắn liền với lịch sử khám phá ra các incretin được nêu ở trên. Như đã biết, các incretin nội sinh, mà chủ yếu là GLP-1 và GIP, đóng vai trò quan trọng trong tạo ra hiệu ứng incretin. DPP-4 là enzyme phân hủy các incretin này. Do vậy như một lẽ tự nhiên, người ta đã nghĩ đến sự phát triển các thuốc có tác dụng ức chế enzyme này để kéo dài tác dụng của các incretin nội sinh, góp phần điều trị đái tháo đường.
Các đại diện trong nhóm: Sitagliptin, Saxagliptin, Vildagliptin, Linagliptin và Alogliptin.
Các thuốc này chỉ làm tăng tiết insulin (không tăng tiết insulin nền) khi nồng độ glucose huyết tương tăng lên, giúp hạ đường huyết sau ăn. Cân nặng ít thay đổi (có thể giảm nhẹ). Không có tác dụng trên độ rỗng dạ dày (không giống như các thuốc chủ vận thụ thể GLP-1).
Các thuốc nhóm này đều hấp thu nhanh qua đường tiêu hóa. Hoạt động ức chế DPP-4 thường kéo dài (cả ngày). Trừ Vildagliptin phải dùng 2 lần/ngày, tất cả các thuốc còn lại đều dùng 1 lần/ngày.
Một số thuốc có chuyển hóa qua gan nên cần chú ý tương tác thuốc. Thải trừ chủ yếu qua thận (trừ Linagliptin thải trừ qua mật) nên có thể cần chỉnh liều ở bệnh nhân suy thận.
Ngoài DPP-4, trong cơ thể còn có một số enzyme khác có cấu trúc tương tự là DPP-8 và DPP-9. Các thuốc nhóm này đều ức chế chọn lọc trên DPP-4 hơn, nhưng mức độ chọn lọc khác nhau (cao nhất là Linagliptin và Alogliptin, thấp nhất là Vildagliptin và Saxagliptin).
Hiệu lực ở mức trung bình, giảm HbA1c 0.7-1.0%.
Thuốc có làm giảm biến chứng vi mạch, tuy nhiên bằng chứng lâm sàng chưa đủ do mới ra đời chưa lâu.
Các thuốc nhóm này hiện nay cũng xuất hiện ngày càng nhiều ở Việt Nam, tuy nhiên rào cản lớn nhất đối với nó vẫn là vấn đề chi phí. Trước đây chúng từng được quảng cáo rất rầm rộ ở Việt Nam, tuy nhiên chúng ta cần cân nhắc thật kỹ về yếu tố chi phí – hiệu quả mà nó đem lại. Không phải cứ thuốc càng đắt tiền thì càng tốt.
Cơ chế tác dụng: Các thuốc nhóm này đều liên kết với trung tâm hoạt động của enzyme, ức chế vị trí xúc tác của DPP-4 bằng cách bắt chước cấu trúc đầu N dipeptide của incretin. Vildagliptin và Saxagliptin có tương tác cộng hóa trị, còn Sitagliptin, Linagliptin và Alogliptin có tương tác không cộng hóa trị.
Chỉ định: Đái tháo đường type 2.
Tác dụng không mong muốn:
- Về lý thuyết, thuốc có thể ảnh hưởng đến hệ miễn dịch. Tuy nhiên trên thực tế, bằng chứng lâm sàng không nhiều. Các tác dụng không mong muốn được báo cáo phổ biến bao gồm đau đầu, viêm mũi họng và nhiễm trùng hô hấp trên.
- Nguy cơ viêm tụy (Tương tự các thuốc chủ vận thụ thể GLP-1): Không dùng các thuốc này cho người có tiền sử hoặc nghi ngờ viêm tụy.
- Nguy cơ tim mạch, bao gồm bệnh mạch vành, suy tim không rõ ràng.
- FDA cảnh báo (2015) về khả năng gây đau khớp nặng của các thuốc nhóm này.
- Alogliptin: Báo cáo rối loạn chức năng gan. Cần xét nghiệm chức năng gan định kỳ khi sử dụng thuốc này (3 tháng 1 lần).
- Phản ứng dị ứng: Phù mạch, phản ứng phản vệ, viêm da tróc vảy, hội chứng Stevens-Johnson (Alogliptin, Sitagliptin và Saxagliptin).
Chống chỉ định: Quá mẫn cảm với bất cứ thành phần nào của thuốc. Suy thận, gan nặng. Phụ nữ mang thai và đang cho con bú.
Thuốc ức chế SGLT2:
Đây có thể nói là các thuốc mới nhất trong điều trị đái tháo đường type 2 được FDA phê duyệt. Tất cả các thuốc trong nhóm này đều có đuôi là “gliflozin”. Các thuốc này hiện nay chưa phổ biến ở Việt Nam và giá thành cũng khá “chát”.
Các đại diện trong nhóm: Dapagliflozin, Canagliflozin, Empagliflozin…
Nguy cơ hạ đường huyết quá mức của các thuốc trong nhóm này thấp hơn Insulin và các sulfonylureas.
Các thuốc nhóm này có hiệu lực trung bình, với mức giảm HbA1c khoảng 0.5-0.7%.
Các thử nghiệm lâm sàng cho thấy các thuốc này có tác dụng giảm cân nhẹ (1-4 kg), giảm huyết áp tâm thu đáng kể. Bệnh nhân dùng các thuốc ức chế SGLT2 không bị hạ natri máu như các thuốc lợi tiểu. Ban đầu người ta cho rằng tác dụng hạ huyết áp có thể do tác dụng lợi tiểu, nhưng sử dụng thuốc lâu dài có thể là do ức chế hệ thống renin-angiotensin-aldosterone (hệ RAA) và giảm cân. Thuốc ức chế SGLT2 có thể là lựa chọn lý tưởng cho các bệnh nhân bị đái tháo đường type 2 và tăng huyết áp.
Các thuốc nhóm này làm giảm nguy cơ biến chứng mạch máu nhỏ, nhưng nguy cơ biến chứng mạch máu lớn (biến cố tim mạch) có giảm hay không lại chưa nhất quán giữa các thử nghiệm lâm sàng.
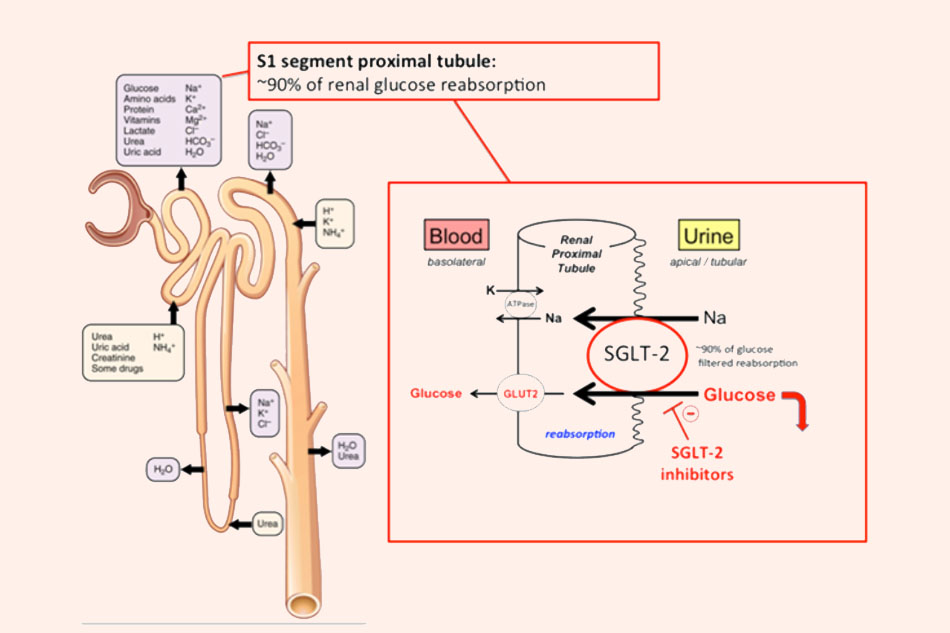
Cơ chế tác dụng: Các thuốc trong nhóm này tập trung ức chế chính vào SGLT2, là một kênh đồng vận chuyển natri và glucose ở ống lượn gần. Kênh này đóng vai trò tái hấp thu tới 90% lượng glucose trong nước tiểu đầu. Khi bị ức chế, glucose không được tái hấp thu và cứ thế đi ra ngoài theo nước tiểu. Điều này giúp hạ glucose máu. Glucose đi vào nước tiểu nhiều làm tăng áp lực thẩm thấu trong lòng ống thận, nó hút nước vào trong lòng ống thận, gây ra đa niệu thẩm thấu.
Cũng đồng thời tồn tại một vấn đề, đó là các thuốc ức chế SGLT2 này không hoàn toàn chọn lọc trên SGLT2, chúng cũng có thể ức chế cả kênh SGLT1. Điều này góp phần vào một số tác dụng không mong muốn của thuốc.
Chỉ định: Đái tháo đường type 2.
Tác dụng không mong muốn:
- Nhiễm trùng tiết niệu – sinh dục (tăng gấp 4 lần trong các thử nghiệm lâm sàng): Nguyên nhân là do trong nước tiểu của bệnh nhân dùng thuốc có nhiều glucose, là môi trường thuận lợi cho vi sinh vật phát triển.
- Kích thích tăng tổng hợp các thể ketone: Do thuốc làm tỷ lệ insulin/glucagon thấp hơn. Tăng nguy cơ nhiễm toan ketone, đặc biệt ở cấc bệnh nhân mắc đái tháo đường type 1.
- Viêm tụy cấp do Canagliflozin (hiếm gặp).
- FDA cảnh báo giảm mật độ xương, tăng nguy cơ gãy xương: Canagliflozin.
Chống chỉ định: Bệnh nhân suy thận (GFR < 45 mL/phút/1.73m2). Bệnh nhân đái tháo đường type 1. Bệnh nhân nhiễm toan ketone.
Amylin analogues:
Amylin là một hormone có cấu trúc polypeptide 37 acid amin được tuyến tụy tiết ra, có liên quan đến quá trình đồng hóa glucose. Nó làm chậm tốc độ tháo rỗng dạ dày và làm giảm sự thèm ăn. Amylin cũng làm ức chế tiết glucagon và hạ glucose huyết tương sau ăn. Tác dụng của nó có nhiều điểm tương đồng với các incretin.
Pramlintide là một thuốc bắt chước cấu trúc của amylin nội sinh trong cơ thể, với vài thay đổi nhỏ trong cấu trúc phân tử.
Về hiệu lực, Pramlintide làm giảm HbA1c không nhiều, khoảng 0.6%. Cân nặng bệnh nhân cũng giảm đáng kể nhưng không nhiều.
Không giống như các thuốc tác dụng kiểu incretin, bao gồm thuốc chủ vận GLP-1 và thuốc ức chế DPP-4, Pramlintide không yêu cầu tế bào β đảo tụy phải còn chức năng để thể hiện tác dụng hạ đường huyết đầy đủ của nó. Đó chính là lý do vì sao nó được sử dụng trong đái tháo đường type 1.
Pramlintide có làm giảm các biến chứng mạch máu nhỏ, nhưng các biến chứng mạch máu lớn thì không.
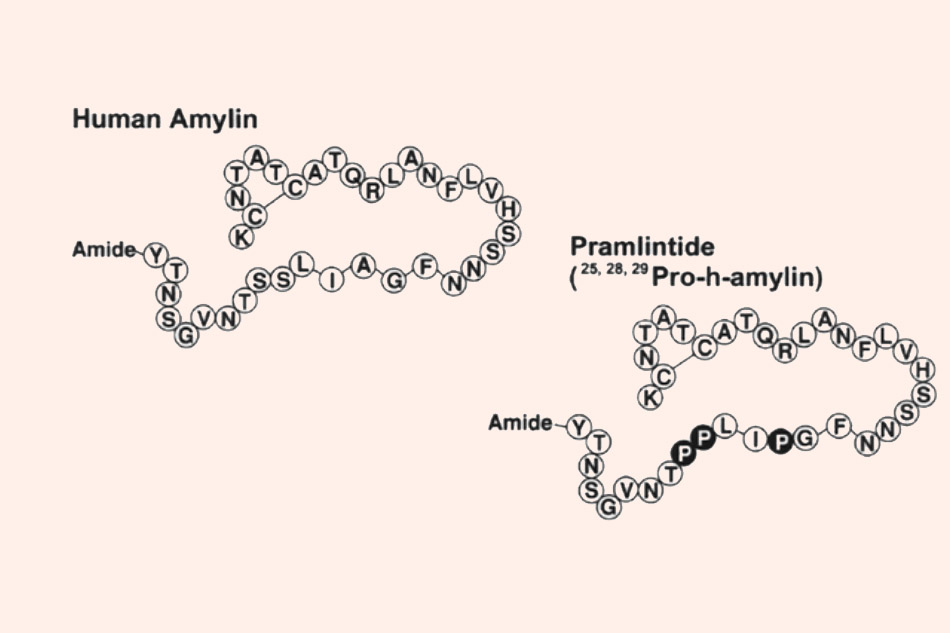
Cơ chế tác dụng: Cơ chế tác dụng của thuốc tương tự amylin nội sinh của người. Pramlintide cũng làm chậm tốc độ tháo rỗng dạ dày và làm giảm sự thèm ăn nên góp phần giảm cân (tuy nhiên cơ chế đầy đủ chưa được hiểu rõ hoàn toàn), giảm bớt tác dụng không mong muốn gây tăng cân của Insulin.
Chỉ định: Đái tháo đường type 1 và type 2 (điều trị bổ sung cho Insulin).
Tác dụng không mong muốn:
- Buồn nôn, nôn, chán ăn, hạ đường huyết quá mức.
- Phản ứng tại chỗ tiêm, dị ứng.
- Viêm tụy.
Chống chỉ định: Quá mẫn cảm với bất cứ thành phần nào của thuốc. Liệt dạ dày. Hạ đường huyết không nhận thức được.
Thuốc ức chế aldose reductase:
Ở những người có lượng đường trong máu bình thường, glucose tế bào chủ yếu đi vào con đường đường phân để tạo ra năng lượng cho cơ thể nhờ được phosphoryl hóa thành glucose 6-phosphate bởi enzyme hexokinase. Chỉ có một lượng rất nhỏ glucose không được phosphoryl hóa (xấp xỉ 3%) đi vào con đường polyol (polyol pathway). Tuy nhiên, ở những bệnh nhân đái tháo đường, do cơ thể khó sử dụng glucose làm năng lượng hơn (do glucose không vào được tế bào), có thể có tới hơn 30% lượng glucose đi vào con đường polyol.
Trong con đường polyol, ban đầu glucose được chuyển thành sorbitol nhờ xúc tác bởi aldose reductase (AR, enzyme khử aldose), đây là bước quyết định tốc độ cả quá trình, có sự tham gia của nicotinamide adenosine dinucleotide phosphate (NADPH), là chất cho proton. Sorbitol sau đó được chuyển thành fructose nhờ sorbitol dehydrogenase (SDH, enzyme dehydrogen hóa sorbitol), có sự tham gia của dạng oxy hóa của nicotinamide adenine dinucleotide (NAD+, cofactor), đây là chất nhận proton. Con đường polyol lần đầu tiên được khám phá ra ở trong túi tinh bởi Hers (1956), nhà khoa học này cũng là người đã chứng minh được glucose trong máu được chuyển thành fructose, tạo thành nguồn năng lượng cho các tế bào tinh trùng. AR đã được tìm thấy trong một số mô của người và động vật, bao gồm nhiều vùng khác nhau ở mắt, cũng như tinh hoàn, buồng trứng, cơ xương, gan, thận, nhau thai, hồng cầu, tim và não.
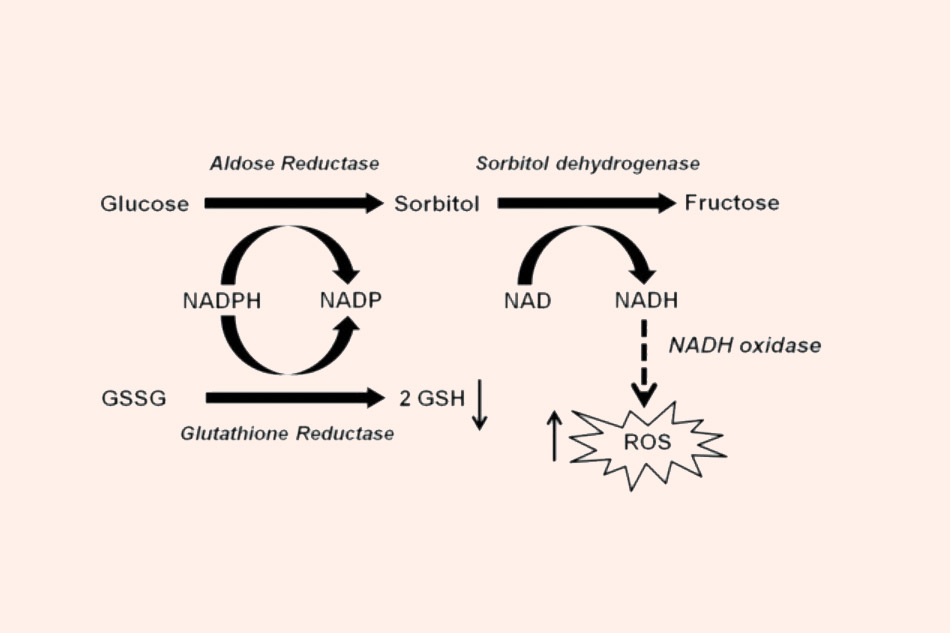
Vai trò của AR trong tạo ra các loại oxy phản ứng (reactive oxidative species, ROS) gây stress oxy hóa ở bệnh nhân đái tháo đường. Lượng glucose dư thừa được chuyển sang con đường polyol, ở đó nó được AR xúc tác, biến thành sorbitol nhờ NADPH. Vì NADPH đóng vai trò rất quan trọng trong việc tạo ra GSH (glutathione, chất chống oxy hóa nội bào) từ GSSG (2 phân tử glutathione được nối với nhau bằng cầu nối disulfide), nên sự giảm nồng độ NADPH nội bào do con đường này gây ra có thể làm giảm khả năng chống oxy hóa nội bào. Sorbitol sau đó được chuyển thành fructose nhờ SDH và tạo ra NADH từ NAD+, NADH dưới tác dụng của NADH oxidase có khả năng làm tăng các ROS, gây stress oxy hóa, làm hại tế bào.
Con đường polyol có liên quan đến các biến chứng vi mạch đái tháo đường. Cơ chế chính được đề xuất cho nó được cho là do qua trung gian thay đổi chuyển hóa và thẩm thấu các mô, tăng sản xuất các ROS gây tổn thương những tế bào và mô nào có con đường này.
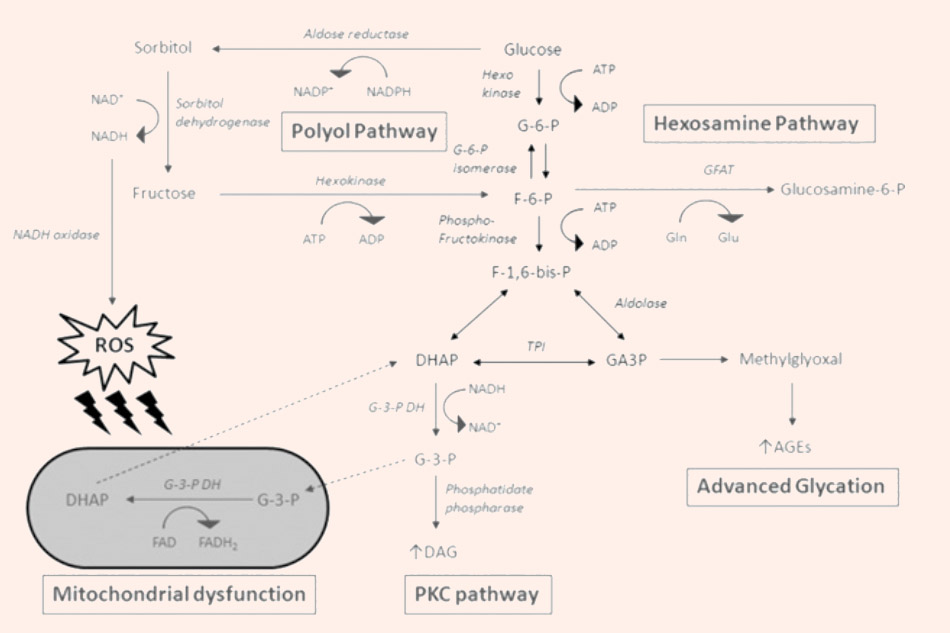
Một số cơ chế tiềm tàng gây ra những tổn thương trong biến chứng vi mạch do đái tháo đường thông qua tăng lượng glucose đi vào con đường polyol. Sorbitol được tăng cường tạo ra ở bệnh nhân đái tháo đường theo con đường polyol, gây ra stress thẩm thấu. Fructose là sản phẩm cuối cùng của con đường polyol, nó được chuyển thành fructose-6-phosphate (F-6-P) nhờ có hexokinase xúc tác, sau đó F-6-P được chuyển thành glucosamine-6-phosphate nhờ enzyme GFAT (glutamine: fructose-6-phosphate amidotransferase) (hexosamine pathway), hoặc cũng có thể nó được chuyển thành fructose-1,6-bisphosphate (F-1,6-P), chất mà sau đó được chuyển thành dihydroxyacetone phosphate (DHAP). DHAP và G3P (glyceraldehyde-3-phosphate) vốn dĩ là 2 đồng phân của nhau và có thể được hỗ biến, chuyển hóa qua lại lẫn nhau nhờ enzyme đồng phân hóa triosephosphate isomerase. G3P có thể tạo ra methylglyoxal, làm tăng nồng độ AGEs. Mặt khác, cũng có thể chuyển DHAP thành diacylglycerol (DAG), làm hoạt hóa con đường PKC (PKC pathway). Sự chuyển dạng liên tục từ glycerol-3-phosphate (G-3-P) thành DHAP trong ty thể dẫn đến sự tạo ra nhiều FADH2, có thể làm tăng điện thế màng ty thể và ức chế chuỗi vận chuyển điện tử ở phức hợp III trong hô hấp tế bào. NADH oxidase thì oxy hóa NADH và tạo ra các loại oxy phản ứng (ROS), có thể tấn công màng ty thể, làm rối loạn chức năng ty thể.
Bên cạnh bệnh mạch máu đái tháo đường, AR cũng được phát hiện có vai trò quan trọng trong bệnh cơ tim đái tháo đường, bệnh này đặc trưng bởi rối loạn chức năng co bóp cơ tim, khác với bệnh động mạch vành đã biết.
Các thuốc trong nhóm ức chế AR này không có tác dụng hạ đường huyết, mà chúng có tác dụng phòng và điều trị các biến chứng mạch máu nhỏ trong đái tháo đường, mà chủ yếu là biến chứng thần kinh.
Đại diện: Epalrestat.
Cơ chế tác dụng: Các thuốc nhóm này ức chế enzyme AR, từ đó làm giảm chuyển hóa glucose theo con đường sorbitol, giảm sự hình thành các loại oxy phản ứng (ROS) cũng như làm tăng nồng độ chất chống oxy hóa nội bào glutathione, bảo vệ tế bào khỏi stress oxy hóa.
Chỉ định: Dự phòng hoặc điều trị các biến chứng thần kinh do đái tháo đường type 1 và type 2.
Tác dụng không mong muốn:
- Rối loạn tiêu hóa: Buồn nôn, nôn, tiêu chảy.
- Đau đầu, chóng mặt.
- Dị ứng.
Chống chỉ định: Quá mẫn cảm với bất cứ thành phần nào của thuốc. Phụ nữ có thai và cho con bú (chống chỉ định tương đối).
Các nhóm thuốc dự phòng biến cố tim mạch
Các thuốc trong nhóm này không có tác dụng hạ đường huyết như nhóm 1, nhưng lại có vai trò cực kỳ quan trọng trong điều trị đái tháo đường vì chúng là các thuốc không thể thiếu giúp giảm thiểu nguy cơ gặp phải các biến cố tim mạch (hay biến chứng mạch máu lớn), giảm nguy cơ nhập viện và tử vong, kéo dài thời gian sống cho bệnh nhân đái tháo đường.
Một số thuốc được sử dụng trong dự phòng các biến cố tim mạch trong đái tháo đường được liệt kê tóm tắt dưới đây.
Thuốc điều trị rối loạn lipid máu:
Thuốc ức chế HMG-CoA reductase:
Các thuốc nhóm này còn được gọi là các “statins”, đây là nhóm thuốc điều trị rối loạn lipid máu được sử dụng phổ biến nhất và cũng là nhóm thuốc quan trọng nhất. Bộ Y tế Việt Nam khuyến cáo sử dụng các statins này để dự phòng các biến cố tim mạch trong đái tháo đường.
Đại diện: Lovastatin, Simvastatin, Atorvastatin, Rosuvastatin, Fluvastatin, Pravastatin.
Các thuốc này đa số có thời gian bán thải ngắn nên phải dùng buổi tối trước khi đi ngủ để đạt nồng độ đỉnh trong huyết tương vào buổi đêm, trùng với khoảng thời gian tổng hợp cholesterol nội sinh trong gan diễn ra mạnh nhất. Thuốc có thời gian bán thải (t¬1/2) dài như Atorvastatin có thể dùng vào buổi sáng.
Nhiều statins được chuyển hóa qua hệ thống CYP450, đặc biệt là CYP3A4. Cần hết sức lưu ý vấn đề tương tác thuốc với các statins.
Để tìm hiểu sâu hơn về các thuốc nhóm này, mời bạn đọc tìm hiểu bài viết Thuốc ức chế HMG-CoA reductase.
Cơ chế tác dụng: Các thuốc nhóm này ức chế theo cơ chế cạnh tranh một enzyme gan có tên HMG-CoA reductase, enzyme chịu trách nhiệm chuyển HMG-CoA thành mevalonate, bước quyết định tổng hợp cholesterol nội sinh trong gan. Khi bị ức chế, cholesterol nội sinh trong gan giảm, gan tăng cường biểu hiện các LDL receptor trên màng tế bào, bắt giữ LDL-C từ máu về gan, điều này làm giảm LDL-C trong máu. VLDL cũng được giảm giải phóng từ gan vào máu nên giảm nhẹ. CÒn HDL-C thì tăng nhẹ.
Các nghiên cứu sau này cũng cho thấy thuốc có tác dụng chống viêm mạch máu nhẹ, bảo vệ chức năng nội mạc mạch máu thông qua chống stress oxy hóa. Điều này có thể cũng góp phần vào làm giảm nguy cơ vữa xơ động mạch.
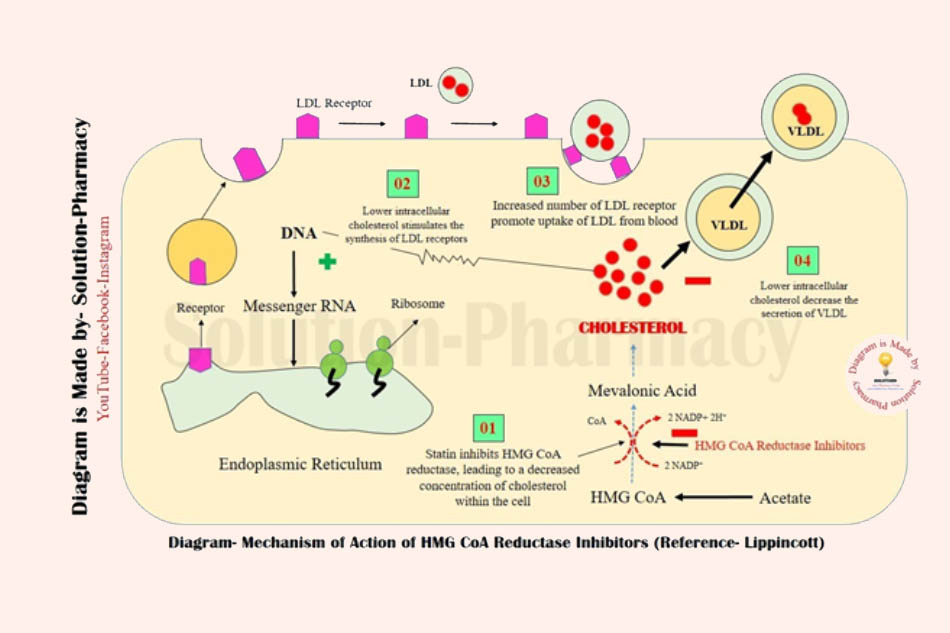
Chỉ định: Rối loạn lipid máu nguyên phát type IIa và IIb theo phân loại của Fredrickson, dự phòng nguyên phát và thứ phát các biến cố tim mạch trong đái tháo đường, bệnh mạch vành, tăng cholesterol máu gia đình hoặc có nguy cơ cao bệnh tim mạch mà không liên quan đến 3 bệnh kể trên.
Việc sử dụng statin nào sẽ phụ thuộc vào hiệu lực của statin và phân tầng nguy cơ tim mạch của bệnh nhân.
Tác dụng không mong muốn:
- Bệnh cơ, yếu cơ và tiêu cơ vân cấp: Nguyên nhân là do statins làm giảm CoQ10 và ATP trong cơ. Nếu tiến triển nặng có thể gây suy thận cấp do tắc nghẽn. Thường có tình trạng tăng creatine kinase (CK) máu. Xử trí các tác dụng không mong muốn liên quan đến bệnh lý cơ vân khá phức tạp.
- Tăng transaminase gan.
- Tăng đường huyết (cảnh báo của FDA).
- Rối loạn tiêu hóa: Buồn nôn, nôn, táo bón, tiêu chảy…
Chống chỉ định: Quá mẫn cảm với bất cứ thành phần nào của thuốc. Phụ nữ có thai và đang cho con bú. Bệnh gan tiến triển, tăng men gan dai dẳng không rõ lý do. Một số phối hợp có thể làm tăng cao nồng độ statins trong máu (ví dụ: Simvastatin + Clarithromycin).
Thuốc chủ vận receptor nhân PPAR-α:
Các thuốc chủ vận receptor nhân PPAR-α còn được gọi là các “fibrates”. Không giống các statins, các fibrates không có chỉ định cho dự phòng các biến cố tim mạch. Nhưng trong trường hợp các statins đơn độc không đủ để đưa mức LDL-C về mục tiêu, các bác sĩ lâm sàng có thể kê đơn phối hợp chúng với statins.
Các đại diện trong nhóm: Fenofibrate, Gemfibrozil, Ciprofibrate, Bezafibrate.
Các thuốc fibrates cũng có tác dụng không mong muốn trên cơ, yếu cơ và tiêu cơ, tác dụng không mong muốn này tăng lên khi phối hợp với statins.
Để biết thêm chi tiết về nhóm thuốc này, mời bạn đọc xem bài viết Thuốc chủ vận receptor nhân PPAR-α.
Cơ chế tác dụng: Thuốc hoạt động theo cơ chế chủ vận một receptor nhân có tên PPAR-α. Điều này dẫn đến điều hòa các gen tham gia vào quá trình chuyển hóa lipid, hoạt hóa lipoprotein lipase, dẫn đến tăng thủy phân triglyceride trong VLDL-C cũng như chylomicron, nên làm giảm VLDL-C và đặc biệt là triglyceride máu. LDL-C giảm nhẹ và HDL-C tăng nhẹ (do tăng tổng hợp Apo A-I và Apo A-II).
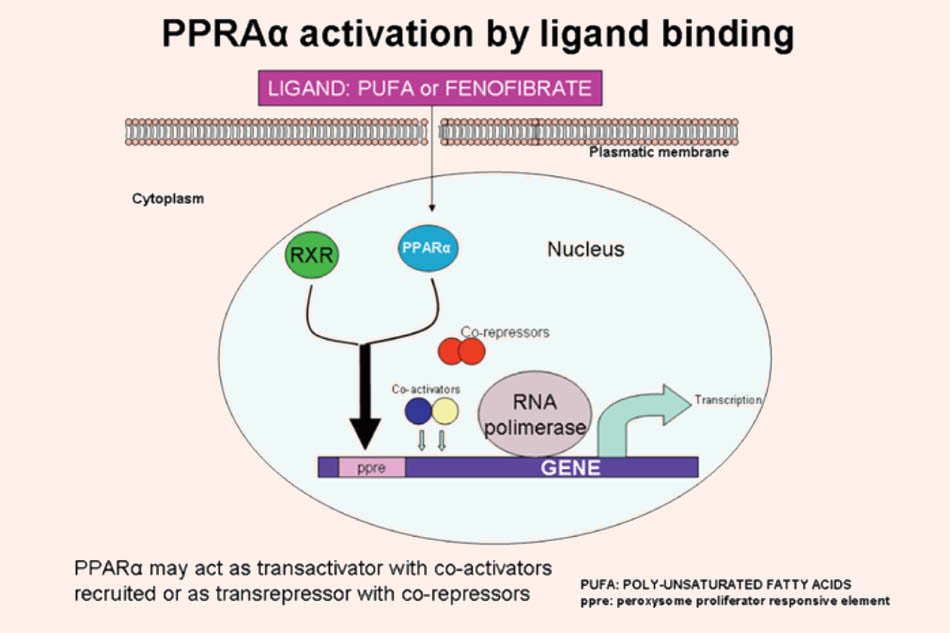
Chỉ định: Rối loạn lipoprotein máu nguyên phát type IIb, III và IV theo phân loại của Fredrickson. Dùng phối hợp với statin khi bệnh nhân chưa kiểm soát tốt lipid máu.
Tác dụng không mong muốn:
- Bệnh cơ, yếu cơ, tiêu cơ vân, đặc biệt khi phối hợp với statins.
- Rối loạn tiêu hóa: Buồn nôn, nôn, tiêu chảy…
- Tăng nguy cơ sỏi mật do bài tiết cholesterol qua mật tăng.
Chống chỉ định: Quá mẫn cảm với bất kỳ thành phần nào của thuốc. Phụ nữ có thai. Bệnh nhân suy gan, thận nặng.
Nhựa gắn acid mật:
Nhựa gắn acid mật còn gọi là các “resins”. Đây là các thuốc cũng khá phổ biến trong điều trị rối loạn lipid máu, nhưng không có chỉ định trong dự phòng các biến cố tim mạch. Ở bệnh nhân đái tháo đường, nó chỉ nên được kết hợp với statins để đưa mức LDL-C về mục tiêu nếu statins đơn độc là không đủ.
Các thuốc nhóm này đều không hấp thu qua đường tiêu hóa và thải trừ dưới dạng gắn với acid mật qua phân.
Đại diện: Cholestyramine, Colestipol.
Cơ chế tác dụng: Các thuốc trong nhóm này tạo phức bền vững với acid mật hoặc muối mật được tiết ra từ gan. Dưới dạng phức bền vững như vậy, acid mật hoặc muối mật không thể được tái hấp thu theo chu kỳ gan – ruột bình thường được, nó bị đào thải ra ngoài theo phân. Điều này phản hồi âm tính trở về gan làm gan tăng tổng hợp acid mật bù trừ. Để tổng hợp được acid mật, gan phải lấy LDL-C từ máu về thông qua điều hòa lên các thụ thể LDL-C trên bề mặt tế bào gan, từ đó làm giảm LDL-C trong máu. Ngoài ra, do acid mật hoặc muối mật bị gắn với resins nên giảm khả năng nhũ hóa lipid, cholesterol được hấp thu từ thức ăn cũng giảm.
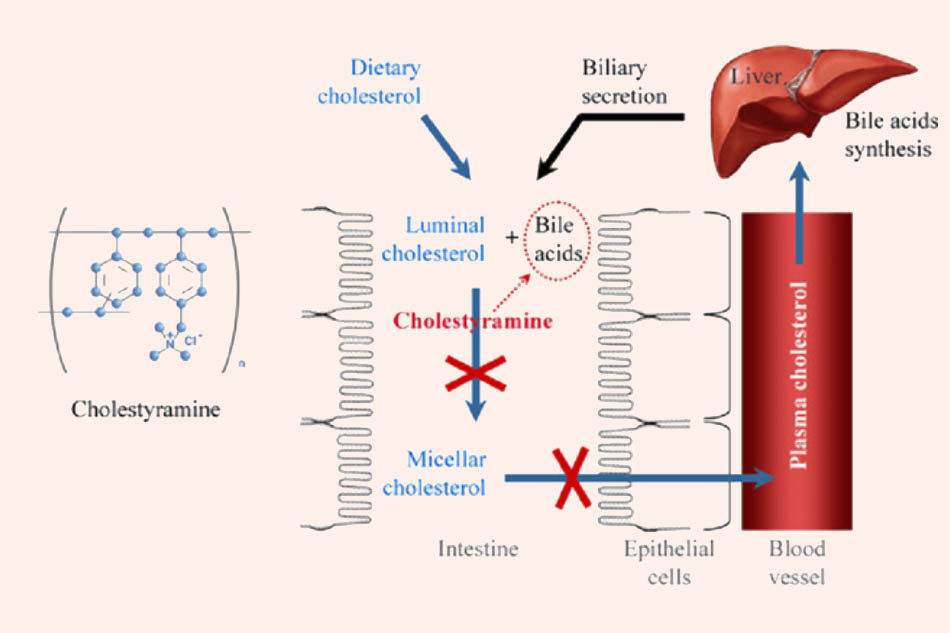
Các thuốc nhóm này làm giảm LDL-C 15-20%, HDL-C ít thay đổi nhưng làm tăng triglyceride huyết tương.
Chỉ định: Rối loạn lipoprotein máu type IIa theo phân loại của Fredrickson. Phối hợp với statins cho phép giảm liều statins, hạn chế các tác dụng không mong muốn.
Tác dụng không mong muốn:
- Rối loạn tiêu hóa: Tiêu chảy.
- Các vitamin tan trong dầu (A, D, E, K) bị giảm hấp thu. Xem xét bổ sung khi điều trị dài ngày.
- Ảnh hưởng đến sự hấp thu một số thuốc khác. Có thể cần dùng các thuốc đó 2 giờ trước hoặc 6 giờ sau khi dùng các resins.
Chống chỉ định: Quá mẫn cảm với bất kỳ thành phần nào của thuốc. Tăng triglyceride máu nặng. Tắc nghẽn đường mật.
Thuốc ức chế hấp thu cholesterol
Đại diện duy nhất cho nhóm này hiện nay được dùng phổ biến là Ezetimibe. Thuốc này không được dùng đơn độc nhiều mà chủ yếu là dùng kết hợp với các statins.
Cơ chế tác dụng: Thuốc ức chế trực tiếp kênh vận chuyển cholesterol ở niêm mạc ruột non (NPC1L1), dẫn đến giảm hấp thu cholesterol từ thức ăn. Gan thích nghi với điều này bằng cách tăng bắt giữ LDL-C từ máu về gan thông qua điều hòa lên các LDL receptor ở màng tế bào gan.
Thuốc làm giảm LDL-C mà ít ảnh hưởng đến triglyceride và HDL-C.
Dẫn chất chuyển hóa (liên hợp glucuronide) có hoạt tính mạnh hơn gấp 400 lần chất mẹ, có chu kỳ gan – ruột nên tác dụng kéo dài.
Thuốc có tác dụng hiệp đồng với các statins.
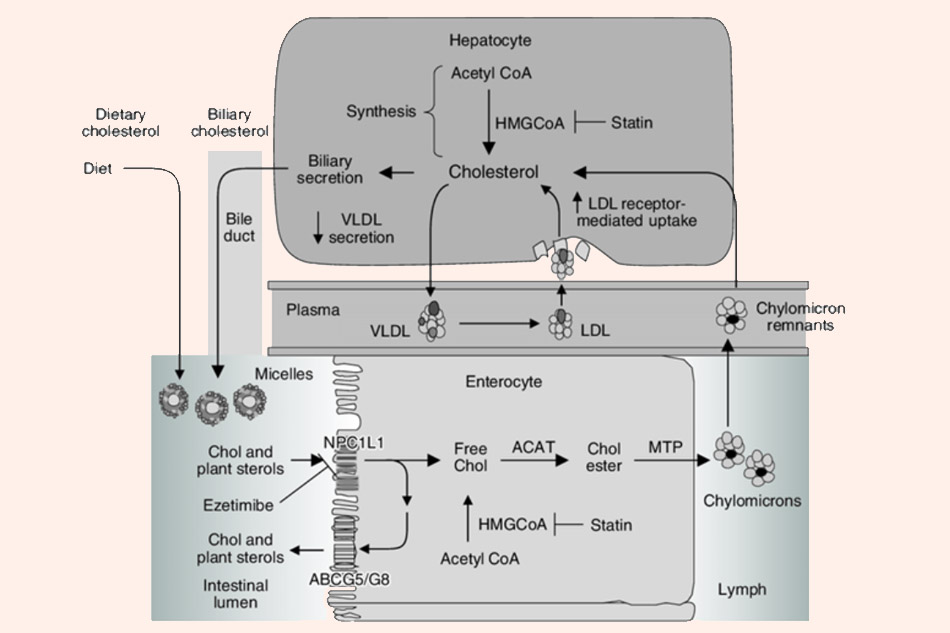
Chỉ định: Phối hợp với statins trong điều trị rối loạn lipoprotein máu type IIa và IIb và dự phòng các biến cố tim mạch.
Tác dụng không mong muốn:
- Triệu chứng kiểu cúm: sổ mũi, đau họng.
- Dùng đồng thời với nhựa gắn acid mật làm giảm sinh khả dụng của thuốc. Nên dùng thuốc 2 giờ trước hoặc 4 giờ sau khi dùng các resins.
Chống chỉ định: Quá mẫn cảm với bất cứ thành phần nào của thuốc. Khi phối hợp với các statins, thuốc sẽ có các chống chỉ định của statins.
Thuốc ức chế kết tập tiểu cầu:
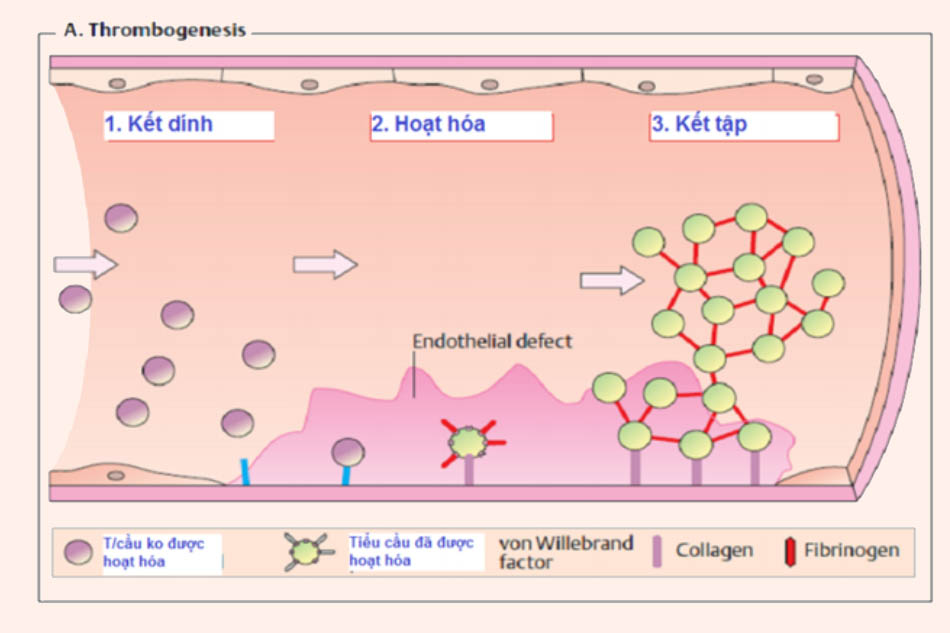
Quá trình kết tập tiểu cầu với 3 giai đoạn: Kết dính, hoạt hóa và kết tập.
Cục máu đông ở động mạch thường giàu tiểu cầu, do đó các thuốc chống kết tập tiểu cầu được sử dụng thường xuyên trong dự phòng các biến cố tim mạch, nguyên phát cũng như thứ phát.
Thuốc ức chế COX-1 trong tiểu cầu:
Đại diện cho nhóm thuốc này là Aspirin.
Dù tất cả các thuốc chống viêm không steroid (NSAIDs) khác đều có tác dụng ức chế kết tập tiểu cầu, nhưng người ta hiện nay chỉ sử dụng Aspirin mà thôi (sẽ làm rõ ở phần cơ chế tác dụng).
Hiện tại có thể nói Aspirin là loại thuốc được sử dụng phổ biến nhất trong số các thuốc ức chế kết tập tiểu cầu, với hiệu lực cao, giá thành rẻ và tương đối ít các tác dụng không mong muốn cũng như tương tác thuốc.
Cơ chế tác dụng: Aspirin ức chế không hồi phục cyclooxygenase-1 (COX-1) của tiểu cầu. Đây là enzyme có tác dụng chuyển acid arachidonic thành prostaglandin H2, từ đó tổng hợp ra thromboxane A2 (TXA2) – tác nhân gây ngưng kết tiểu cầu. Sự ức chế không hồi phục này chính là sự khác biệt giữa Aspirin với các thuốc NSAIDs khác.
Vì ức chế nhanh và không hồi phục COX-1 nên tiểu cầu nào đã bị ức chế COX-1 thì coi như mất khả năng kết tập. Cơ thể cần thay thế nó bằng tiểu cầu mới (đời sống trung bình của tiểu cầu là 7-10 ngày). Dùng Aspirin liều lặp lại sẽ tạo tác dụng tích lũy trên tiểu cầu.
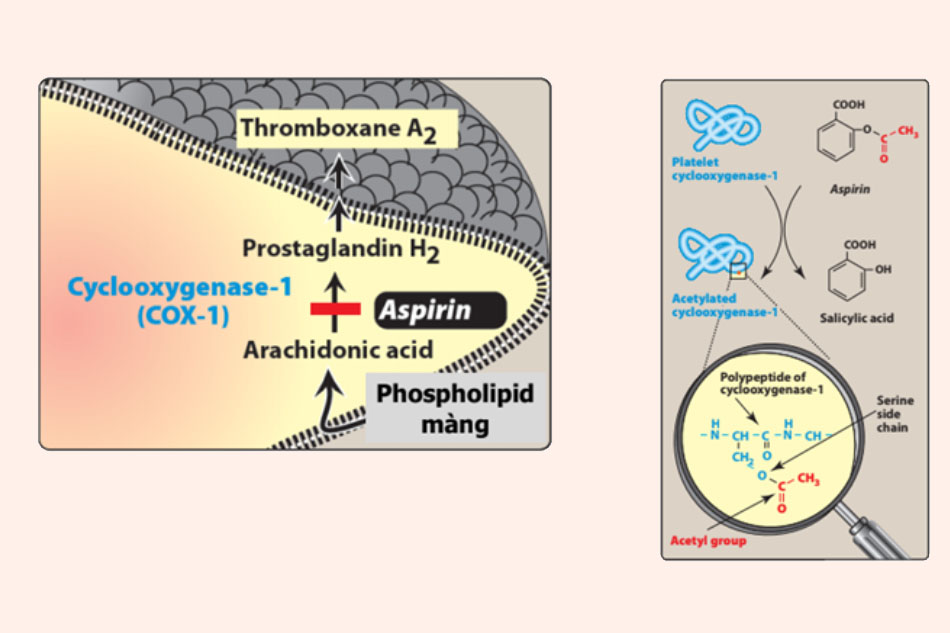
Đây là cơ chế tác dụng của Aspirin. Trái: Aspirin ức chế hình thành TXA2. Phải: Aspirin acetyl hóa COX-1, làm nó mất hoạt tính vĩnh viễn.
Thuốc có tác dụng ức chế kết tập tiểu cầu tốt ở liều không quá 325 mg, vì khi đó nó chưa ảnh hưởng nhiều đến prostacyclin (yếu tố chống ngưng kết tiểu cầu ở nội mạc). Ở liều cao hơn, từ 500 mg trở lên, tác dụng chống kết tập tiểu cầu giảm do ức chế cả sự tổng hợp prostacyclin nội mạc mạch máu. Trên thực tế người ta thường dùng Aspirin có hàm lượng 75-100 mg.
Aspirin là chất ức chế không chọn lọc COX. Nó ức chế cả COX-1 và COX-2.
Chỉ định: Dự phòng huyết khối động mạch nguyên phát trên bệnh nhân có nguy cơ cao hoặc thứ phát sau nhồi máu cơ tim, đặt stent, phẫu thuật bắc cầu chủ vành…
Tác dụng không mong muốn:
- Kéo dài thời gian chảy máu: Tác dụng không mong muốn này không thể tránh được và cần hạn chế các hoạt động có thể gây chảy máu.
- Rối loạn tiêu hóa: Khó tiêu, buồn nôn, nôn. Có thể khắc phục bằng uống sau ăn, dùng viên bao tan trong ruột. Liều Aspirin 75-100 mg ít gây loét đường tiêu hóa.
- Ảnh hưởng đến chức năng thận không đáng kể.
- Sử dụng với rượu, corticoids làm tăng nguy cơ loét đường tiêu hóa.
Chống chỉ định: Quá mẫn cảm với Aspirin hoặc bất cứ thành phần nào của thuốc. Bệnh nhân có nguy cơ chảy máu đường tiêu hóa. Không dùng cho trẻ em dưới 12 tuổi (có thể gây ra một tình trạng nghiêm trọng gọi là hội chứng Reye).
Thuốc ức chế P2Y12:
Các đại diện của các thuốc nhóm này là Ticlopidine, Clopidogrel, Prasugrel… Đây cũng là các thuốc được sử dụng phổ biến tại Việt Nam (Clopidogrel thông dụng hơn Prasugrel vì giá thành rẻ hơn, Ticlopidine thì có nhiều tác dụng không mong muốn), thường sử dụng kết hợp với Aspirin để dự phòng các biến cố tim mạch.
Cơ chế tác dụng: Các thuốc nhóm này ức chế kết tập tiểu cầu bằng cách ức chế thụ thể P2Y12 của ADP, khi ADP không gắn được vào thụ thể và từ đó cũng không thể hoạt hóa được GP IIb/IIIa ở sau, tiểu cầu không kết tập được.
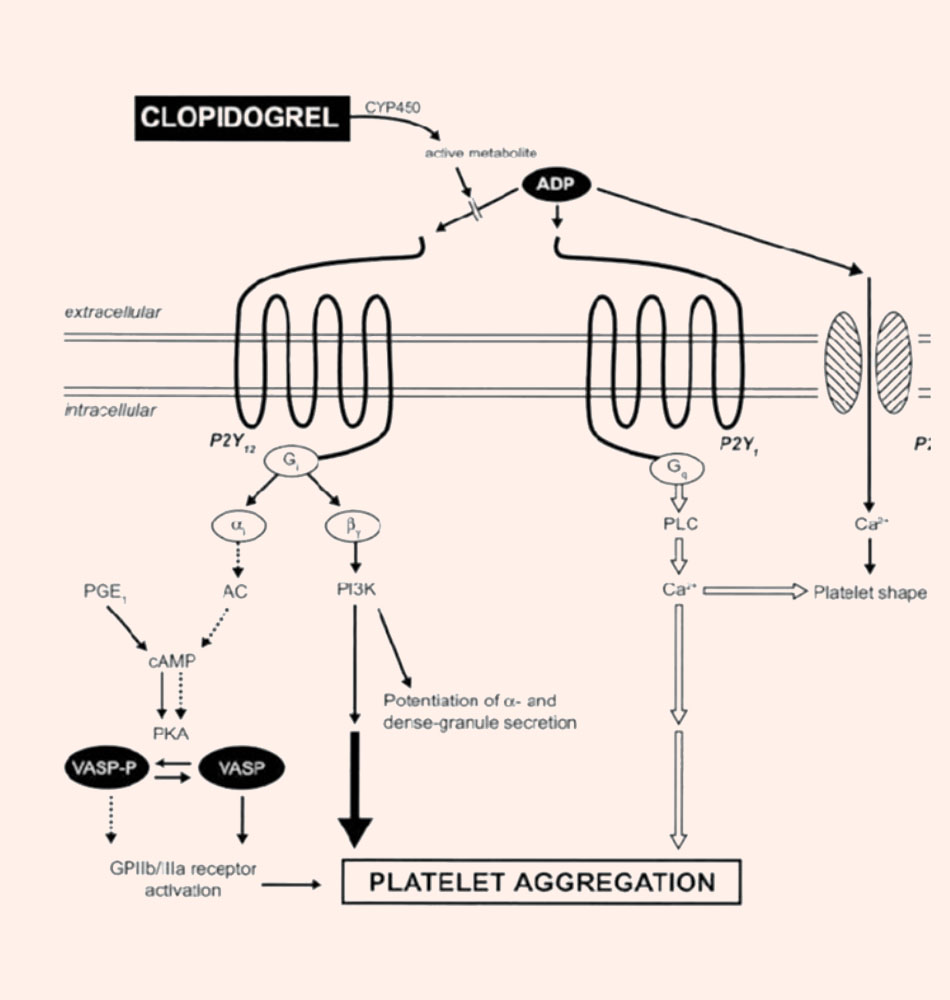
Tác dụng của thuốc phát huy nhanh và được kéo dài liên tục 7-10 ngày.
Chỉ định: Phối hợp với Aspirin để dự phòng nguyên phát hoặc thứ phát các biến cố tim mạch.
Tác dụng không mong muốn:
- Kéo dài thời gian chảy máu: Không thể tránh được và cần tránh để bị chảy máu.
- Clopidogrel là tiền thuốc, phải được chuyển hóa qua CYP2C19 thành chất có hoạt tính. Các thuốc ức chế CYP2C19 (đặc biệt là nhiều thuốc ức chế bơm proton – PPIs) có thể làm giảm tác dụng của Clopidogrel.
- Ticlopidine có nhiều tác dụng không mong muốn hơn: Giảm bạch cầu hạt, giảm tiểu cầu.
Chống chỉ định: Quá mẫn cảm với bất cứ thành phần nào của thuốc. Bệnh nhân chảy máu bệnh lý tích cực bao gồm chảy máu đường tiêu hóa và chảy máu nội sọ.
Thuốc ức chế phosphodiesterase:
Đại diện tiêu biểu của nhóm này là Dipyridamole. Thuốc này hiện tại không được dùng nhiều tại Việt Nam so với Aspirin và Clopidogrel.
Cơ chế tác dụng: Thuốc ức chế enzyme phosphodiesterase tiểu cầu, làm tăng AMP vòng (cAMP) trong tiểu cầu, đồng thời ức chế sự sáp nhập của adenosine vào tiểu cầu và nội mạc, do đó gây ức chế kết tập tiểu cầu. Ngoài ra thuốc cũng có tác dụng giãn mạch.
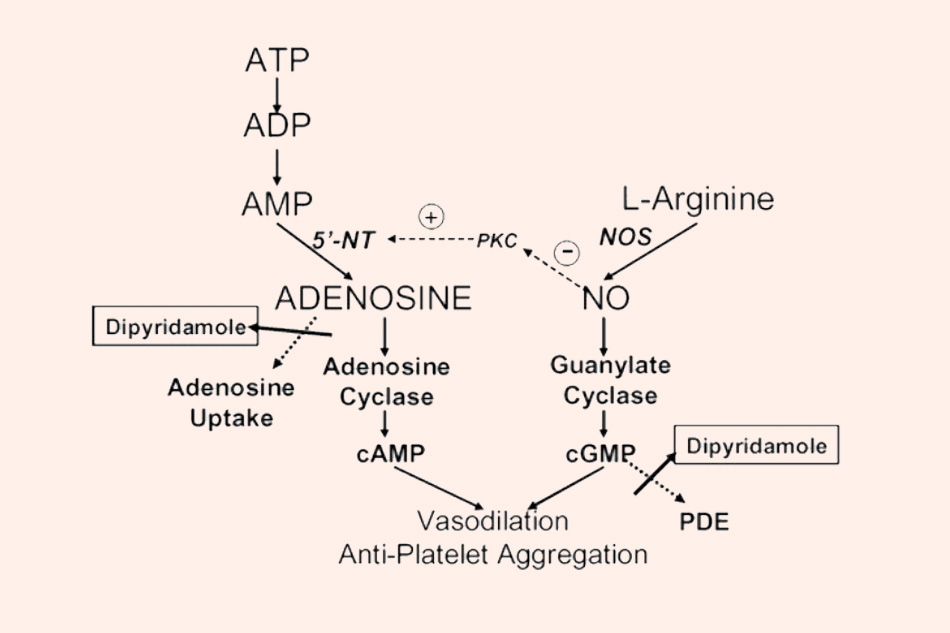
Chỉ định: Dự phòng nguyên phát hoặc thứ phát các biến cố tim mạch. Tuy nhiên hiện nay đây không phải là loại thuốc được dùng nhiều trong dự phòng các biến cố tim mạch ở bệnh nhân đái tháo đường.
Tác dụng không mong muốn:
- Kéo dài thời gian chảy máu: Đây là điều không thể tránh khỏi và nên giáo dục bệnh nhân tránh các hoạt động có thể gây chảy máu.
- Tụt huyết áp, tăng nhịp tim: Có liên quan đến tác dụng giãn mạch.
- Đau đầu, chóng mặt.
- Rối loạn tiêu hóa: Nôn, buồn nôn.
- Dị ứng.
Chống chỉ định: Quá mẫn cảm với bất kỳ thành phần nào của thuốc. Sốc tuần hoàn, trụy mạch.
Thuốc đối kháng thụ thể GP IIb/IIIa:
Thuốc đối kháng thụ thể GP IIb/IIIa là các thuốc ức chế kết tập tiểu cầu mới nhất hiện nay. Vì thế chúng có giá thành không hề rẻ và ở Việt Nam chũng vẫn chưa được phổ biến rộng rãi. Và đi kèm với chi phí điều trị cao thì các thuốc này cũng cho thấy hiệu lực vượt trội của chúng trong các thử nghiệm lâm sàng.
Các thuốc thuộc nhóm này đều là các thuốc dùng đường tĩnh mạch, vì vậy chỉ sử dụng thuốc trong điều trị nội trú chứ không sử dụng trong điều trị ngoại trú.
Cơ chế tác dụng: Khi mô bị tổn thương, tiểu cầu được hoạt hóa và thay đổi hình dạng tại vị trí thụ thể GP IIb/IIIa, cho phép nó liên kết với fibrinogen và yếu tố von Willebrand. Ngoài ra, trên 1 tiểu cầu có nhiều thụ thể GP IIb/IIIa, làm cho các tiểu cầu liên kết chéo với nhau, đồng thời sự tồn tại của cơ chế feedback dương tính gây ra khuếch đại lũy tiến sự hoạt hóa và kết tập tiểu cầu. Các thuốc trong nhóm này đều đối kháng thụ thể GP IIb/IIIa, ngăn chặn bước cuối cùng trong kết tập tiểu cầu. Hiệu lực ức chế kết tập tiểu cầu của các thuốc nhóm này cao hơn Aspirin (có hoặc không có Clopidogrel).
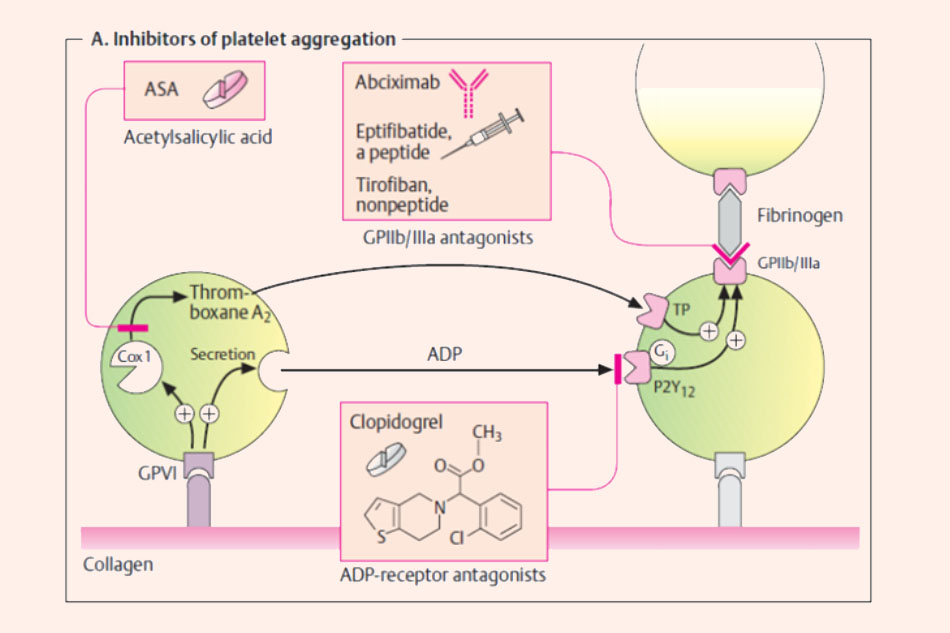
Các thuốc ức chế GP IIb/IIIa có thể thấy trong hình: Abciximab, Eptifibatide, Tirofiban.
Ngoài tác dụng ức chế sự kết tập tiểu cầu, thuốc đối kháng thụ thể GP IIb/IIIa còn có khả năng gây ra sự hòa tan cục máu đông giàu tiểu cầu bằng cách phá vỡ sự tương tác giữa tiểu cầu và fibrinogen.
Chỉ định: Dự phòng thứ phát các biến cố tim mạch. Thường chúng ta không dùng thuốc này dự phòng nguyên phát vì sự bất tiện trong đường dùng của nó. Với dự phòng thứ phát, cũng chỉ sử dụng thuốc trong mổ thời gian ngắn điều trị nội trú ban đầu, khi bệnh nhân không thể sử dụng các thuốc đường uống.
Tác dụng không mong muốn:
- Kéo dài thời gian chảy máu, dễ xuất huyết: Tác dụng không mong muốn này phụ thuộc liều và cần được bác sĩ lâm sàng quản lý chặt chẽ.
- Rối loạn tiêu hóa: Nôn, buồn nôn.
- Đau đầu, hạ huyết áp.
- Phản ứng đau tại chỗ tiêm.
Chống chỉ định: Quá mẫn cảm với bất cứ thành phần nào của thuốc. Bệnh nhân có xuất huyết hoạt động, giảm tiểu cầu. Loét dạ dày. Phẫu thuật chấn thương gần đây, khối u nội sọ, tăng huyết áp không kiểm soát, viêm mạch.
Thuốc điều trị tăng huyết áp:
Thuốc ức chế hệ RAA:
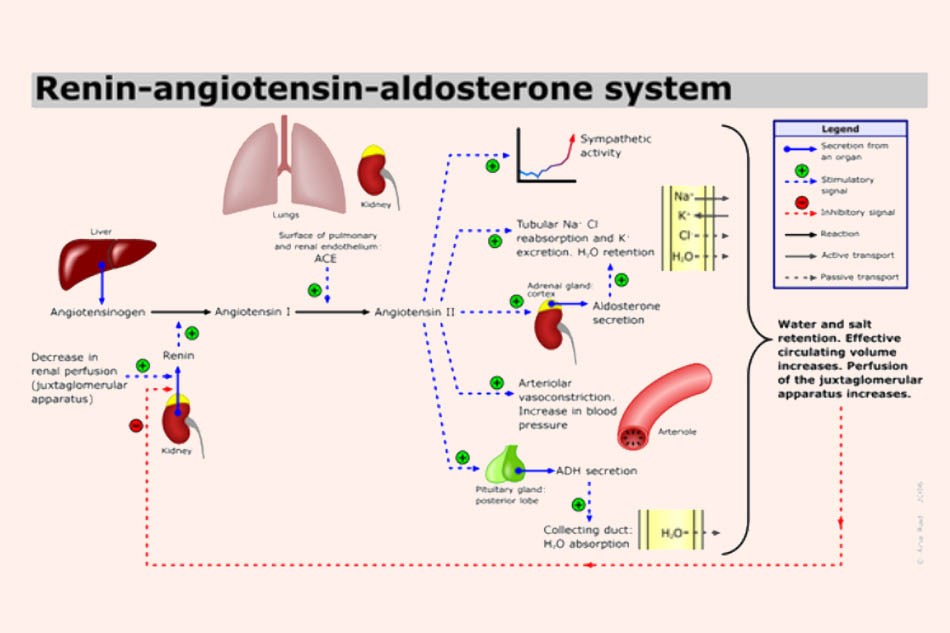
Các thuốc ức chế hệ renin-angiotensin-aldosterone (RAA) đóng một vai trò quan trọng không chỉ trong điều trị tăng huyết áp nói chung mà còn có vai trò quan trọng với bệnh nhân đái tháo đường có tăng huyết áp. Đây là các thuốc được chỉ định đầu tiên khi bệnh nhân đái tháo đường có tăng huyết áp, đặc biệt là khi có đi kèm với tình trạng protein niệu.
Khi vi một nguyên nhân nào đó mà mức lọc cầu thận giảm, cơ thể sẽ khởi động hệ RAA để duy trì mức lọc cầu thận. Đầu tiên các tế bào cận cầu thận tiết ra renin. Chất này có tác dụng xúc tác chuyển angiotensinogen vốn có sẵn trong máu thành angiotensin I. Angiotensin I sau đó đến phổi và được men ACE ở đây xúc tác chuyển tiếp thành angiotensin II, đây mới là chất có hoạt tính. Nó tác động lên các thụ thể AT, mà ở đây quan trọng nhất trong tăng huyết áp là thụ thể AT1. Angiotensin II gây ra một loạt các tác dụng: co mạch, tăng tái hấp thu natri và nước, tăng tiết aldosterone, tăng tiết ADH gây giữ muối và nước, hoạt hóa hệ thần kinh giao cảm, các điều này làm tăng huyết áp; kích thích phì đại thất trái, sẽ làm nặng hơn tình trạng suy tim trái.
Các thuốc ức chế hệ RAA đều tác động vào một khâu nào đó trong quá trình này, từ đó làm mất tác dụng của angiotensin II.
Thuốc ức chế renin trực tiếp:
Thuốc duy nhất trong nhóm này được phê duyệt đó là Aliskiren. Thuốc này hiện tại chưa được phổ biến ở Việt Nam.
Thuốc này có sinh khả dụng đường uống khá thấp, chỉ 2.5%. Thuốc được chuyển hóa qua hệ thống enzyme gan CYP450 3A4, nhưng mức độ thì chưa rõ, cần chú ý tương tác thuốc qua hệ enzyme này. Liên kết với protein huyết tương khoảng 50% và thể tích phân bố (Vd) khoảng 135 L. Thời gian bán hủy (t1/2) dài (20-45 giờ).
Cơ chế tác dụng: Aliskiren là một non-peptide phân tử lượng thấp, ức chế cạnh tranh renin để nó không chuyển được angiotensinogen thành angiotensin I, từ đó làm mất tác dụng của angiotensin II và gây hạ huyết áp.
Chỉ định: Tăng huyết áp.
- Tác dụng không mong muốn:
- Hạ huyết áp quá mức.
- Đau đầu, chóng mặt và mệt mỏi.
- Tăng kali máu: do ức chế tiết aldosterone.
Rối loạn tiêu hóa: tiêu chảy.
Chống chỉ định: Quá mẫn cảm với bất cứ thành phần nào của thuốc. Phụ nữ có thai. Trẻ em dưới 2 tuổi. Phối hợp với thuốc ức chế hệ RAA khác.
Thuốc ức chế men chuyển angiotensin (ACEIs):
Các thuốc này là các thuốc được dùng phổ biến nhất trong nhóm ức chế hệ RAA vì hiệu lực tốt và giá thành phải chăng. Các thuốc nhóm này đều có đuôi “pril”.
Các đại diện trong nhóm (phân loại theo cấu trúc):
- Phân nhóm 1 (chứa nhóm sulfuhydryl -SH): Captopril.
- Phân nhóm 2 (chứa 2 nhóm carboxyl, hầu hết dưới dạng ester hóa): Enalapril, Benazepril, Quinapril, Ramipril, Lisinopril, Perindopril…
- Phân nhóm 3 (chứa phospho): Fosinopril.
Các thuốc có cấu trúc ester hóa ở phân nhóm 2 đều là tiền thuốc, khi vào cơ thể, dưới tác dụng của enzyme esterase, các phân tử thuốc bị thủy phân thành dạng có hoạt tính. Người ta cần thiết kết công thức các phân tử thuốc này dưới dạng ester để tăng tính thân lipid, tăng sinh khả dụng của thuốc. Trừ trường hợp đặc biệt là Lisinopril, thuốc này không ở dạng ester mà ở dạng dicarboxylic trực tiếp nhưng vẫn được hấp thu tốt vì thuốc bắt chước cấu trúc của một acid amin tự nhiên là L-Lysine. Ở ruột, Lisinopril được hấp thu chủ yếu theo cơ chế vận chuyển tích cực nhờ hệ vận chuyển các acid amin của ruột non.
Hầu như các thuốc này đều dùng với liều khá thấp (trừ Captopril).
Cơ chế tác dụng: Thuốc ức chế men chuyển angiotensin (ACE) ở phối, ngăn cản nó xúc tác chuyển angiotensin I thành angiotensin II, từ đó tạo ra tác dụng hạ huyết áp do mất tác dụng của angiotensin II.
ACE là enzyme chịu trách nhiệm giáng hóa bradykinin. Ức chế men ACE làm bradykinin không bị giáng hóa, bradykinin cũng gây giãn mạch qua trung gian NO (nitric oxide) và góp phần làm hạ huyết áp.
Các ACEIs đều có thể gây hạ huyết áp liều đầu.
Chỉ định: Tăng huyết áp. Được chỉ định trong đái tháo đường có tăng huyết áp và đặc biệt khi có protein niệu.
Tác dụng không mong muốn:
- Hạ huyết áp quá mức.
- Captopril: Do có nhóm –SH trong cấu trúc nên thuốc này có thể gây ban đỏ, lưỡi vị kim loại.
- Ho: Do bradykinin không được giáng hóa, tích lũy và gây ho. Ho này không đáp ứng với các thuốc giảm ho. Nếu ho quá nhiều, bác sĩ nên xem xét cho bệnh nhân chuyển sang dùng các thuốc chẹn thụ thể angiotensin (ARBs).
- Tăng kali máu: Thuốc cũng làm giảm tiết aldosterone nên làm tăng kali máu.
- Dị ứng: Phù Quincke (hiếm gặp).
Chống chỉ định: Quá mẫn cảm với bất cứ thành phần nào của thuốc. Phụ nữ có thai. Phối hợp với thuốc ức chế hệ RAA khác. Hẹp động mạch thận hai bên.
Thuốc chẹn thụ thể angiotensin (ARBs):
Các thuốc trong nhóm này thường được gọi là các “sartans”. Các thuốc nhóm này tuy có cơ chế khác các ACEIs một chút, nhưng nhìn chung thì các tác dụng, tác dụng không mong muốn và chống chỉ định y hệt nhau. Giá thành các loại thuốc này thường đắt hơn ACEIs, nên không được ưu tiên bằng.
Đại diện: Losartan, Valsartan, Candesartan, Irbesartan…
Nhìn chung các sartans ít có điểm chung. Chúng cùng hấp thu nhanh qua đường tiêu hóa, nhưng sinh khả dụng khác nhau. Phân bố, chuyển hóa và thải trừ cũng đa dạng.
Cơ chế tác dụng: Các thuốc nhóm này bắt chước cấu trúc không gian của angiotensin II và ức chế cạnh tranh thụ thể AT1 của angiotensin II, từ đó làm angiotensin II không thể hiện được tác dụng của nó.
Thuốc không ảnh hưởng đến giáng hóa bradykinin nên ít gây ho hơn các ACEIs. Ngoài ra, không giống như ACEIs, các sartans hạ huyết áp từ từ nên không có hiện tượng hạ huyết áp liều đầu như ACEIs.
Chỉ định: Giống các ACEIs.
Tác dụng không mong muốn:
- Tăng kali máu: Tương tự các ACEIs.
- Dị ứng: Tương tự các ACEIs.
- Hạ huyết áp quá mức: Ít xảy ra khi dùng đơn độc, nhưng dễ xảy ra ở bệnh nhân mất nước (như dùng thuốc lợi tiểu).
Chống chỉ định: Giống các ACEIs.
Thuốc chẹn kênh calcium (CCBs):
Các thuốc chẹn kênh calcium được dùng trong điều trị tăng huyết áp, đặc biệt khi có bệnh thận mạn (có thể là biến chứng của đái tháo đường) do thuốc khá an toàn với thận.
Thường dùng các thuốc thuộc nhóm dẫn chất dihydropyridine (DHP) cho bệnh nhân đái tháo đường vì các thuốc này ít ảnh hưởng đến tim hơn các nhóm dẫn chất phenylalkylamine và benzothiazepine. Dưới đây ta sẽ chỉ nói về các thuốc thuộc nhóm dẫn chất DHP.
Đại diện: Nifedipine, Amlodipine, Nicardipine, Felodipine…
Các thuốc nhóm này nhìn chung hấp thu tốt qua đường tiêu hóa nhưng sinh khả dụng (F) thấp do bị chuyển hóa bước 1 ở gan. Phần lớn liên kết mạnh với protein huyết tương và có thời gian bán thải (t¬1/2) ngắn, thải trừ chủ yếu qua thận dưới dạng không còn hoạt tính (một phần qua phân). Một số thuốc trong nhóm có chuyển hóa qua hệ enzyme gan CYP450 nên cần chú ý tương tác thuốc.
Cơ chế tác dụng: Các thuốc dẫn chất DHP trong nhóm này ngăn không cho ion calcium đi vào nội bào các tế bào cơ trơn động mạch thông qua ức chế các kênh calcium type L trên màng tế bào (bình thường nồng độ ion calcium ngoại bào cao hơn nội bào). Ion calcium cần thiết cho quá trình co cơ, việc ức chế các kênh ion này làm cho cơ trơn động mạch giãn, từ đó hạ huyết áp.
Đây là cơ chế hoạt động của các thuốc chẹn kênh calcium. Ca2+ không thể vào được tế bào nên không liên kết được với calmodulin để hoạt hóa MLCK (myosin light chain kinase). MLCK không được hoạt hóa nên sợi myosin không trượt trên sợi actin để gây co cơ được.
Chỉ định: Tăng huyết áp.
Tác dụng không mong muốn:
- Hạ huyết áp tư quá mức gây nhịp tim nhanh phản xạ, có thể làm nặng thêm thiếu máu cơ tim ở những bệnh nhân có thiếu máu cơ tim cục bộ. Amlodipine ít gây tăng huyết áp phản xạ hơn Nifedipine.
- Rối loạn tiêu hóa: Buồn nôn, ợ nóng, táo bón hoặc tiêu chảy.
- Đau đầu, chóng mặt, đỏ bừng mặt, phù chi dưới, phù phổi do giãn mạch quá mức.
Chống chỉ định: Quá mẫn cảm với bất cứ thành phần nào của thuốc. Huyết áp tâm thu dưới 100 mmHg. Người suy gan nặng.
Thuốc lợi tiểu (Diuretics):
Các thuốc lợi tiểu làm tăng thể tích nước tiểu, giảm thể tích dịch ngoại bào và thể tích tuần hoàn nên hiệp đồng tác dụng hạ huyết áp với các thuốc đã nêu trên.
Lợi tiểu thiazides được lựa chọn phối hợp với các thuốc ACEIs hoặc ARBs do hiệp đồng dược lực học, mặc dù nó cũng có những tác dụng bất lợi với bệnh nhân đái tháo đường, do:
- Hai thuốc này làm giảm bớt tác dụng không mong muốn của nhau trên kali máu: Thuốc ức chế hệ RAA làm tăng kali máu, thiazides làm giảm kali máu.
- Thiazides làm hoạt hóa hệ RAA, nhưng khi phối hợp với các thuốc ức chế hệ RAA sẽ không phải lo về vấn đề này.
Ngoài ra, các thử nghiệm lâm sàng đã chỉ ra kết quả có lợi của việc phối hợp các thuốc ức chế hệ RAA với lợi tiểu thiazides trên bệnh nhân đái tháo đường có tăng huyết áp.
Dưới đây chúng ta sẽ chỉ nói về lợi tiểu thiazides.
Tác dụng lợi tiểu của các thiazides ở mức độ trung bình. Thiazides còn được gọi là lợi tiểu trần thấp (đối lập với Furosemide còn được gọi là lợi tiểu trần cao). Liều thiazides thường thấp và không thể tăng nhiều.
Cơ chế tác dụng: Các thiazides ức chế kênh đồng vận chuyển Na+/Cl- ở đoạn đầu ống lượn xa, làm cho Na+ bị thải ra ngoài theo đường nước tiểu, kéo theo nước gây lợi tiểu. Liều cao còn ức chế men CA (carbonic anhydrase) gây lợi tiểu nhưng đây chỉ là cơ chế thứ yếu.
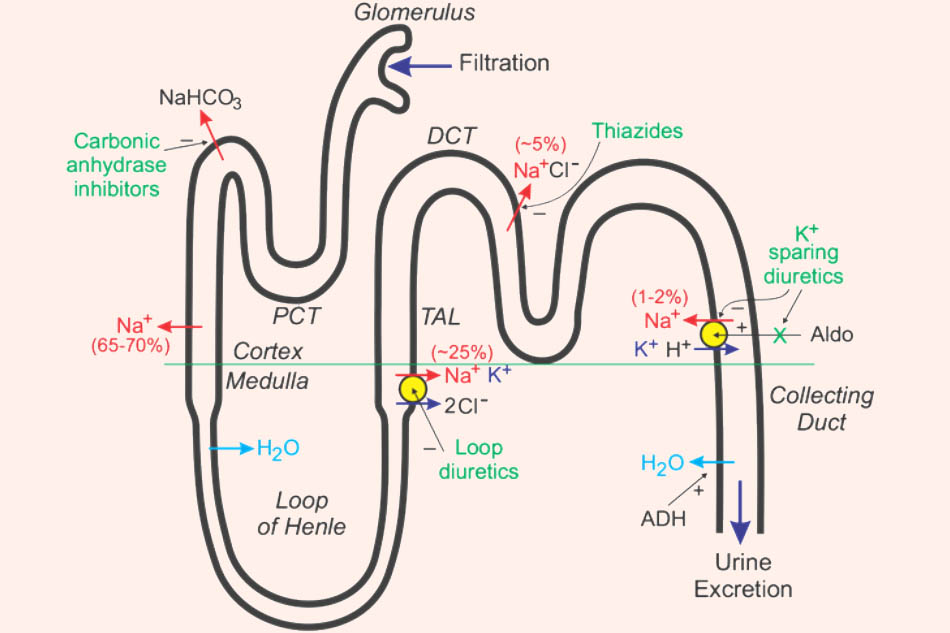
Lợi tiểu ức chế CA: ống lượn gần, lợi tiểu quai: nhánh lên quai Henle, lợi tiểu thiazides: đoạn đầu ống lượn xa, lợi tiểu tiết kiệm kali: đoạn cuối ống lượn xa và đầu ống góp.
Ngoài cơ chế lợi tiểu để hạ huyết áp, thiazides cũng hạ huyết áp theo cơ chế ức chế tác dụng của các chất như vasopressin, noradrenaline (norepinephrine), vốn là các chất gây co mạch.
Chỉ định: Tăng huyết áp. Phù (do bệnh lý tim, gan, thận). Thuốc cũng được dùng trong hạ calcium máu, nhiễm toan chuyển hóa.
Tác dụng không mong muốn:
- Nhiễm kiềm chuyển hóa: Do không tái hấp thu được ion Cl- nên tăng tái hấp thu HCO3- thay thế.
- Hạ kali máu, hạ natri máu. Nguyên nhân hạ kali máu là do nồng độ natri trong nước tiểu cao, cơ thể đáp ứng bằng cách tăng thải kali để kéo natri lại.
- Dùng lâu: Tăng calcium máu, giảm magnesi máu.
- Tăng acid uric máu nên tạo điều kiện thuận lợi cho bệnh gout phát triển.
- Ức chế giải phóng insulin, tăng tiết các catecholamine nên làm tăng glucose máu, tăng cholesterol máu. Đây là các tác dụng bất lợi với bệnh nhân đái tháo đường.
- Dị ứng.
Chống chỉ định: Quá mẫn cảm với bất cứ thành phần nào của thuốc. Bệnh nhân xơ gan có hạ kali máu. Bệnh nhân đang dùng glycoside tim (như Digoxin). Người suy gan, suy thận, bệnh gout. Dị ứng với các sulfamide kháng khuẩn.
Xem thêm:
- [Guideline 2019] Hướng dẫn ESC 2019 Đái tháo đường, tiền đái tháo đường, và bệnh tim mạch
- Bệnh đái tháo đường type 2: Nguyên nhân, biến chứng & phác đồ điều trị




{kind=link}